хромато-масс-спектрометрия
- хромато-масс-спектрометрия
Слитно или раздельно? Орфографический словарь-справочник. — М.: Русский язык.
.
1998.
Смотреть что такое «хромато-масс-спектрометрия» в других словарях:
-
хромато-масс-спектрометрия — chromatografinė masių spektrometrija statusas T sritis Standartizacija ir metrologija apibrėžtis Chromatografiniu būdu atskirtų medžiagų masių spektrometrija atitikmenys: angl. chromatographic mass spectrometry rus. хромато масс спектрометрия, f … Penkiakalbis aiškinamasis metrologijos terminų žodynas
-
хромато-масс-спектрометрия — chromatografinė masių spektrometrija statusas T sritis chemija apibrėžtis Chromatografiniu būdu atskirtų medžiagų masių spektrometrija. atitikmenys: angl. chromatographic mass spectrometry rus. хромато масс спектрометрия … Chemijos terminų aiškinamasis žodynas
-
ХРОМАТО-МАСС-СПЕКТРОМЕТРИЯ — метод анализа смесей гл. обр. орг. в в и определения следовых кол в в в в объеме жидкости. Метод основан на комбинации двух самостоят. методов хроматографии и масс спектрометрии. С помощью первого осуществляют разделение смеси на компоненты, с… … Химическая энциклопедия
-
ХРОМАТО-МАСС-СПЕКТРОМЕТРИЯ — метод анализа смесей гл. обр. органич. соединений. В основе Х. м. с. лежит колоночная газовая (или жидкостная) хроматография и масс спектрометрия. Посредством первого метода осуществляется разделение смеси на отдельные компоненты, с помощью… … Естествознание. Энциклопедический словарь
-
МАСС-СПЕКТРОМЕТРИЯ — (масс спектроскопия, масс спектральный анализ), метод анализа в ва путем определения массы (чаще, отношения массы к заряду m/z) и относит. кол ва ионов, получаемых при ионизации исследуемого в ва или уже присутствующих в изучаемой смеси.… … Химическая энциклопедия
-
Масс-спектрометрия — (масс спектроскопия, масс спектрография, масс спектральный анализ, масс спектрометрический анализ) метод исследования вещества путём определения отношения … Википедия
-
Масс-спектрометр — Масс спектрометрия (масс спектроскопия, масс спектрография, масс спектральный анализ, масс спектрометрический анализ) метод исследования вещества путём определения отношения массы к заряду (качества) и количества заряженных частиц, образующихся… … Википедия
-
Масс-спектроскопия — Масс спектрометрия (масс спектроскопия, масс спектрография, масс спектральный анализ, масс спектрометрический анализ) метод исследования вещества путём определения отношения массы к заряду (качества) и количества заряженных частиц, образующихся… … Википедия
-
Чувствительность масс-спектроскопии — У этого термина существуют и другие значения, см. Чувствительность. Чувствительность в масс спектроскопии величина, показывающая какое количество вещества нужно ввести в масс спектрометр для того, чтобы его можно было детектировать. Для простоты… … Википедия
-
Метаболомика — Метаболомика это «систематическое изучение уникальных химических „отпечатков пальцев“ специфичных для процессов, протекающих в живых клетках» конкретнее, изучение их низкомолекулярных метаболических профилей.[1] Метаболом представляет … Википедия

Хроматограмма по полному ионному току образца воды (а); масс-хроматограмма по току ионов с отношениями массы ионов к их заряду m/z 141 (оранжевый) и 156 (зелёный), характерными соответственно для мети…
ХРОМА́ТО-МАСС-СПЕКТРОМЕТРИ́Я (хроматомасс-спектрометрия, ХМС), комбинированный метод прямого качественного и количественного химич. анализа сложных смесей, сочетающий хроматографич. разделение веществ с их масс-спектрометрическим анализом. Первый вариант ХМС – совмещение газового хроматографа и масс-спектрометра (ГХ/МС) – осуществлён в 1957. Метод ГХ/МС широко применяется и в 21 в., хотя термин «хромато-масс-спектрометрия» сегодня включает жидкостную хроматографию/масс-спектрометрию (ЖХ/МС), суперкритическую флюидную хроматографию/масс-спектрометрию (СФХ/МС), ионную хроматографию/масс-спектрометрию (ИХ/МС), капиллярный электрофорез/масс-спектрометрию (КЭ/МС). ХМС даёт возможность анализа смесей, состоящих из тысяч химич. соединений, и используется для разл. аналитов: от неорганич. ионов до сложнейших биополимеров, включая белки, углеводы, нуклеиновые кислоты.
Анализируемая смесь вводится в хроматограф, где её компоненты разделяются и поочерёдно поступают в масс-спектрометр. Ионизация, разделение образовавшихся ионов и их регистрация дают возможность получить масс-спектры. Результат эксперимента включает хроматограмму образца и масс-спектр каждого компонента. По ним можно провести идентификацию (качественный анализ) и оценить количество компонентов в исходном образце (количественный анализ).
Существует 2 варианта использования ХМС. Первый связан с детектированием целевых соединений, когда задача заключается в количественном определении заранее выбранных аналитов, а все остальные компоненты образца игнорируются. Второй – т. н. скрининг или анализ полного спектра – существенно более сложная задача, включающая идентификацию всех компонентов образца.
Популярный режим ХМС – масс-хроматография – заключается в проведении эксперимента с получением полных масс-спектров, по которым проводят идентификацию. Для количественного определения компьютер строит хроматограммы по току характеристических для каждого соединения ионов. Внешний вид хроматограммы может полностью измениться по сравнению с исходной, поскольку все ионы, за исключением заранее выбранных, игнорируются. Сигналов целевых аналитов может не быть на хроматограмме по полному ионному току, а на масс-хроматограмме они будут интенсивными (рис.). Измерение точных масс ионов (масс-спектрометрия высокого разрешения) создаёт условия для увеличения надёжности результатов ХМС.
Если задачей анализа является количественное определение конкретных микрокомпонентов, для повышения чувствительности используют масс-фрагментографию (мониторинг заданных ионов, селективное ионное детектирование), когда масс-спектрометр настроен на регистрацию характеристических ионов с заданными значениями m/z. Этот метод чувствительнее на два порядка и эффективен для суперэкотоксикантов (напр., полихлорированных дибензодиоксинов). Выигрыш в чувствительности сопровождается значит. проигрышем в информативности и надёжности.
Новейшим вариантом масс-фрагментографии для определения целевых аналитов является тандемная ХМС в режиме мониторинга заданных реакций, когда регистрируют фрагментацию выбранного иона-предшественника (обычно молекулярного иона) с образованием известного иона-продукта. Селективность определения повышается, поскольку фрагментный характеристический ион образуется из характеристического иона-предшественника. Один из самых надёжных методов ХМС – одновременная регистрация двух реакций фрагментации иона-предшественника с образованием наиболее интенсивного сигнала иона-продукта для количественного определения и второго по интенсивности – для подтверждения идентификации аналита.
ХМС – информативный, надёжный и чувствительный аналитич. метод. В варианте ГХ/МС в прибор необходимо ввести лишь неск. фемтограммов соединения. В случае ЖХ/МС при работе с пептидами и белками регистрируются аттомолярные концентрации аналитов. ХМС эффективна не только в фундам. исследованиях, но и для решения практич. задач химич. пром-сти, биологии и медицины (напр., диагностика заболеваний), энергетики (установление состава нефтей и качества топлив), допинг-контроля. Метод незаменим в экологии, криминалистике, антитеррористической деятельности и мн. др. областях.


04.10.2022
Хромато-масс-спектрометрия — это гибридный метод анализа, сочетает хроматографию и масс-спектрометрию. При этом хроматографию необходимо разделять на жидкостную (ВЭЖХ) и газовую (ГХ) т.к. возможны оба варианта. Данное сочетание усиливает возможности обоих методов в результате химик получает уникальный аналитический комплекс. Хроматография получает высокочувствительный детектор, универсальный и селективный одновременно, с уникальной способностью по идентификации компонентов. Возможности МСД для качественного анализа в хроматографии превосходят возможности любых других детекторов. Детектор МСД позволяет идентифицировать соединения не только по временам удерживания, но и сравнивая масс-спектр пика (определяемого вещества) с библиотечным. Библиотеки насчитывают сотни тысяч различных соединений, а программное обеспечение проводит поиск в считанные секунды. Масс-спектрометр благодаря хроматографу сканирует индивидуальные соединения таким образом аналитик работает с чистыми масс-спектрами. Без хроматографии даже при сканировании чистых веществ спектр включает все спектры примесей входящих в исследуемое вещество соединений.
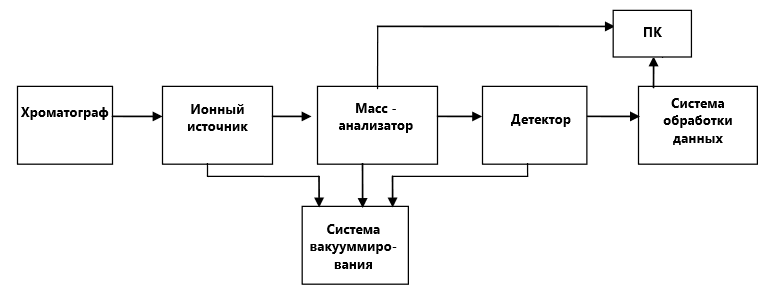
Схема хромато-масс-спектрометра.
Любой хромато-масс-спектрометр можно разделить на следующие блоки:
- Хроматограф. Разделение введенной смеси веществ на индивидуальные компоненты.
- Ионный источник превращает определяемые вещества и заряженные ионы.
- Разделение ионов в соответствии их массой посредством электрических или магнитных полей.
- Детектирование ионов
- Обработка полученных данных
- Достаточно высокий вакуум поддерживается в масс-анализаторе и детекторе, в некоторых системах и в ионном источнике.
Масс-спектрометры, применяемые в ГХ и ВЭЖХ отличаются конструктивно. В первую очередь по принципу ионизации. Разберем основные варианты.
Ионизация в ГХ/МС.
Наиболее старый и наиболее широко применяемый в современной масс-спектрометрии метод ионизации молекул – это электронный удар или электронная ионизация (ЭИ или EI). Именно этот вариант является наиболее распространенным в работе ГХ/МС. Суть электронной ионизации: определяемые вещества в газообразном состоянии из хроматографической колонки поступают в камеру источника ионов, где подвергаются бомбардировке электронами, испускаемыми катодом (филаментом). Катод (филамент) — это металлическая спираль (проволока) из тугоплавкого металла. Излучение электронов происходит при нагреве катода до высоких температур за счет пропускания через него электрического тока, внешне можно сравнить с лампой накаливания. За счет термоэлектронной эмиссии нагретая проволока испускает электроны. Два магнита, расположенные выше и ниже источника ионов, образуют магнитное поле. Под действием магнитного поля электроны движутся по спирали, таким образом, увеличивается длина траектории движения и соответственно увеличивается эффективность ионизации нейтральных молекул. Электроны, испускаемые катодом в ионизационную камеру, ускоряются под действием электрического поля между катодом и ионизационной камерой. Катод поддерживается при отрицательном потенциале относительно ионизационной камеры, величина потенциала регулируется, но обычно равна минус 70 В, что соответствует энергии электронов 70 эВ.
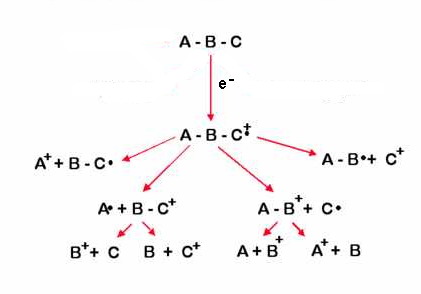
От энергии электронов зависит механизм ионизации молекул. Чем выше энергия, тем сильнее разбивается молекула на более мелкие осколки. 70 эВ – это стандарт, именно с этой энергией ионизации снято большинство масс-спектров входящих в стандартные библиотеки. Мы так подробно остановились на электронной ионизации, потому что это основной вариант для ГХ/МС.
Второй возможный, но менее распространенный вариант ионизации — это химическая ионизация (ХИ или CI). При этом способе источник ионов заполняется каким-либо газом (обычно метан или изобутан, очень редко аммиак и другие газы), который ионизуется все тем же электронным ударом, а в результате большой популяции молекул в источнике начинают происходить ионно-молекулярные реакции, ведущие к образованию ионов-реагентов, которые, в свою очередь взаимодействуют с молекулами интересующего нас вещества, ведя к их ионизации. Такая ионизация является «мягкой», то есть образовавшиеся ионы не разваливаются на мелкие фрагменты, а скорее остаются крупными кусками либо чуть меньше, чем исходная молекула, либо даже большее ее за счет присоединения других ионов. Этот метод дает меньше информации о том, как устроена структура молекулы, зато с его помощью легче определить ее молекулярную массу.
Итак, для ГХ/МС характерны следующие 2 типа ионизации:
- Электронная ионизация (EI)
- Химическая ионизация (CI)
Ионизация в ВЭЖХ/МС
Очень многие органические вещества невозможно испарить без разложения, а это значит, что их нельзя ионизовать электронным ударом. Все это очень характерно для соединений, которые анализируются методами ВЭЖХ, поэтому здесь применяют другие подходы для ионизации.
- ионизация в электроспрее (ESI)
- химическая ионизация при атмосферном давлении (APCI)
- фотоионизаця при атмосферном давлении (APPI)
- ионизация лазерной десорбцией при содействии матрицы (MALDI)
В первом случае (электроспрей) жидкость (интересующие нас соединения с растворителем) вырывается под давлением вместе с коаксиально подаваемым разогретым газом (азотом) из узкого капилляра (иглы, которая находится под повышенным потенциалом – 5-10 кВ) с огромной скоростью и прямо в этой струе мелкодисперсного тумана с оболочек молекул срываются электроны, превращая их в ионы. Большая часть растворителя при движении этой струи переходит в газовую фазу и не попадает в отверстие входного конуса источника ионов API. В режиме химической ионизации при атмосферном давлении потенциал прикладывается не к игле, через которую поступает жидкость, а к электроду в области распыления, что приводит к образованию коронного разряда. В этом случае фрагментация значительно меньше, чем в предыдущем – ESI. В методе MALDI лазерный луч вырывает ионы с поверхности мишени, на которую нанесен образец со специально подобранной матрицей.
Способы разделения ионов в МСД
После того как проведена ионизация образца, необходимо разделить образовавшиеся ионы. Для той цели используется несколько типов анализаторов которые можно разделить на 2 типа: непрерывные и пульсовые.
Непрерывные масс-анализаторы:
- Магнитные. Разделение ионов происходит за счет использования однородного секторного поперечного магнитного поля. Такие спектрометры отличаются высоким разрешением, чувствительностью и широким диапазоном детектируемых масс. Однако они имеют большие габариты и высокую стоимость.
- Квадрупольные. Ионный пучок проходит между четырьмя параллельно и симметрично расположенными электродами, на которые попарно подается определённая комбинация постоянного и высокочастотного напряжения. Под действием небольшого ускоряющего напряжения (7 – 15 В), приложенного к стержням, ионы из источника ионов влетают вдоль общей оси стержней и под действием комбинации постоянного и высокочастотного (радиочастотного) поля, создаваемого стержнями, начинают колебаться в поперечном направлении. При этом амплитуда колебаний ионов возрастает без изменения их направления движения. Ионы, чьи амплитуды колебаний достигают высоких значений, нейтрализуются при столкновении со стержнями, а системы детектирования достигнут только те ионы, чьи значения m/z будут отвечать определенному соотношению значений постоянного и высокочастотного напряжений, приложенных к стержням. Путем изменения значений величин постоянного и высокочастотного напряжений во времени в масс-фильтре создаются условия для прохождения к системе детектирования ионов с определенным диапазоном масс. Таким образом, осуществляется сканирование и получается масс-спектр в определенном диапазоне масс. Приборы имеют компактные размеры, высокую чувствительность и быстродействие. Верхний предел пропускания отношений m/z как правило не превышает 1200 а.е.м. Квадрупольные хромато-масс-спектрометры являются наиболее доступными по цене.
Пульсовые масс-анализаторы:
- Времяпролетный. В основе работы прибора положена зависимость скорости движения заряженных частиц от их массы. Движение ионов в устройстве происходит в бесполевом пространстве. К достоинствам относится практически неограниченный диапазон масс и очень быстрое время регистрации масс-спектра. Часто используются для исследования проб, которые трудно перевести в газовую фазу.
- Ионно-циклотронного резонанса с Фурье-преобразованием. Здесь ион движется под действием сразу двух полей: сильного постоянного магнитного и переменного электрического. Под действием магнитного поля ион движется по окружности с циклической частотой определяемой массой иона и магнитной индукцией. Зашумленные сигналы для сохранения полезной информации подвергаются математическому преобразованию Фурье. Такие приборы обеспечивают высокое разрешение и широкий диапазон измеряемых масс. Для их работы требуется создание сильного магнитного поля.
- Ионная ловушка. В спектрометре предусмотрена две пары электродов: кольцевые и концевые. Для сбора и удержания ионов в полости ловушки используется комбинация постоянного и высокочастотного напряжения. Резонансная радиочастота обеспечивает доступ заряженных частиц на детектор в соответствии с величиной m/z. Ионизации пробы осуществляется с использованием электронного или химического способа. Благодаря селективной регистрации ионов чувствительность прибора значительно повышается.
Вакуумная система
Масс-спектрометр требует создания в нем очень чистого вакуума. Давление остаточного газа в приборе обычно составляет около 10-7 – 10-10 мм.рт.ст. Для создания такого вакуума применяются, как правило системы состоящие из двух насосов: форвакуумного (например, пластинчато-роторного) и высоковакуумного (например, диффузионного или турбомолекулярного). Высокий вакуум необходим для создания пространства без молекул с которыми бы могли сталкиваться анализируемые ионы и нейтрализоваться, не долетая до детектора. Во всех масс-спектрометрах вакуумируется масс-анализатор и система детектирования. В МСД для газовой хроматографии вакуумируется еще и система ионизации. В ВЭЖХ вакуумирование системы ионизации зависит от типа ионизации.
Система детектирования.
Третья обязательная деталь масс-спектрометра – регистрирующее устройство, с помощью которого можно определить количество ионов с данным m/z. Первые масс-спектрографы использовали в качестве детектора фотопластинку. Сейчас используются динодные вторично-электронные умножители, в которых ион, попадая на первый динод, выбивает из него пучок электронов, которые в свою очередь, попадая на следующий динод, выбивают из него еще большее количество электронов и т.д. Другой вариант – фотоумножители, регистрирующие свечение, возникающее при бомбардировке ионами люминофора. Кроме того, используются микроканальные умножители, системы типа диодных матриц и коллекторы, собирающие все ионы, попавшие в данную точку пространства (коллекторы Фарадея). В современном приборе регистрирующее устройство непосредственно связано с компьютером, который производит обработку результатов и управляет экспериментом.
Хромато-масс-спектрометр – это сложный инструмент с широкими возможностями. Компания Хроматограф.ру предлагает газовые хромато-масс-спектрометры Кристалл 5000 под задачи Заказчика. Комплектация подбирается строго под нужды Заказчика. Наши специалисты помогут подобрать нужный Вам хромато-масс-спектрометр. Кроме этого, сервисные инженеры Хроматограф.ру выполняют техническое обслуживание, запуск, ремонт хроматографов c масс-детекторами Agilent 5977B.

Example of a GC-MS instrument
Gas chromatography–mass spectrometry (GC-MS) is an analytical method that combines the features of gas-chromatography and mass spectrometry to identify different substances within a test sample.[1] Applications of GC-MS include drug detection, fire investigation, environmental analysis, explosives investigation, and identification of unknown samples, including that of material samples obtained from planet Mars during probe missions as early as the 1970s. GC-MS can also be used in airport security to detect substances in luggage or on human beings. Additionally, it can identify trace elements in materials that were previously thought to have disintegrated beyond identification. Like liquid chromatography–mass spectrometry, it allows analysis and detection even of tiny amounts of a substance.[2]
GC-MS has been regarded as a «gold standard» for forensic substance identification because it is used to perform a 100% specific test, which positively identifies the presence of a particular substance. A nonspecific test merely indicates that any of several in a category of substances is present. Although a nonspecific test could statistically suggest the identity of the substance, this could lead to false positive identification. However, the high temperatures (300°C) used in the GC-MS injection port (and oven) can result in thermal degradation of injected molecules,[3] thus resulting in the measurement of degradation products instead of the actual molecule(s) of interest.
History[edit]
The first on-line coupling of gas chromatography to a mass spectrometer was reported in the late 1950s.[4][5] An interest in coupling the methods had been suggested as early as December 1954.[6]
The development of affordable and miniaturized computers has helped in the simplification of the use of this instrument, as well as allowed great improvements in the amount of time it takes to analyze a sample. In 1964, Electronic Associates, Inc. (EAI), a leading U.S. supplier of analog computers, began development of a computer controlled quadrupole mass spectrometer under the direction of Robert E. Finnigan.[7] By 1966 Finnigan and collaborator Mike Uthe’s EAI division had sold over 500 quadrupole residual gas-analyzer instruments.[7] In 1967, Finnigan left EAI to form the Finnigan Instrument Corporation along with Roger Sant, T. Z. Chou, Michael Story, Lloyd Friedman, and William Fies.[8] In early 1968, they delivered the first prototype quadrupole GC/MS instruments to Stanford and Purdue University.[7] When Finnigan Instrument Corporation was acquired by Thermo Instrument Systems (later Thermo Fisher Scientific) in 1990, it was considered «the world’s leading manufacturer of mass spectrometers».[9]
Instrumentation[edit]

The insides of the GC-MS, with the column of the gas chromatograph in the oven on the right.
The GC-MS is composed of two major building blocks: the gas chromatograph and the mass spectrometer. The gas chromatograph utilizes a capillary column whose properties regarding molecule separation depend on the column’s dimensions (length, diameter, film thickness) as well as the phase properties (e.g. 5% phenyl polysiloxane). The difference in the chemical properties between different molecules in a mixture and their relative affinity for the stationary phase of the column will promote separation of the molecules as the sample travels the length of the column. The molecules are retained by the column and then elute (come off) from the column at different times (called the retention time), and this allows the mass spectrometer downstream to capture, ionize, accelerate, deflect, and detect the ionized molecules separately. The mass spectrometer does this by breaking each molecule into ionized fragments and detecting these fragments using their mass-to-charge ratio.

These two components, used together, allow a much finer degree of substance identification than either unit used separately. It is not possible to make an accurate identification of a particular molecule by gas chromatography or mass spectrometry alone. The mass spectrometry process normally requires a very pure sample while gas chromatography using a traditional detector (e.g. Flame ionization detector) cannot differentiate between multiple molecules that happen to take the same amount of time to travel through the column (i.e. have the same retention time), which results in two or more molecules that co-elute. Sometimes two different molecules can also have a similar pattern of ionized fragments in a mass spectrometer (mass spectrum). Combining the two processes reduces the possibility of error, as it is extremely unlikely that two different molecules will behave in the same way in both a gas chromatograph and a mass spectrometer. Therefore, when an identifying mass spectrum appears at a characteristic retention time in a GC-MS analysis, it typically increases certainty that the analyte of interest is in the sample.
Purge and trap GC-MS[edit]
For the analysis of volatile compounds, a purge and trap (P&T) concentrator system may be used to introduce samples. The target analytes are extracted by mixing the sample with water and purge with inert gas (e.g. Nitrogen gas) into an airtight chamber, this is known as purging or sparging. The volatile compounds move into the headspace above the water and are drawn along a pressure gradient (caused by the introduction of the purge gas) out of the chamber. The volatile compounds are drawn along a heated line onto a ‘trap’. The trap is a column of adsorbent material at ambient temperature that holds the compounds by returning them to the liquid phase. The trap is then heated and the sample compounds are introduced to the GC-MS column via a volatiles interface, which is a split inlet system. P&T GC-MS is particularly suited to volatile organic compounds (VOCs) and BTEX compounds (aromatic compounds associated with petroleum).[10]
A faster alternative is the «purge-closed loop» system. In this system the inert gas is bubbled through the water until the concentrations of organic compounds in the vapor phase are at equilibrium with concentrations in the aqueous phase. The gas phase is then analysed directly.[11]
Types of mass spectrometer detectors[edit]
The most common type of mass spectrometer (MS) associated with a gas chromatograph (GC) is the quadrupole mass spectrometer, sometimes referred to by the Hewlett-Packard (now Agilent) trade name «Mass Selective Detector» (MSD). Another relatively common detector is the ion trap mass spectrometer. Additionally one may find a magnetic sector mass spectrometer, however these particular instruments are expensive and bulky and not typically found in high-throughput service laboratories. Other detectors may be encountered such as time of flight (TOF), tandem quadrupoles (MS-MS) (see below), or in the case of an ion trap MSn where n indicates the number mass spectrometry stages.
GC-tandem MS[edit]
When a second phase of mass fragmentation is added, for example using a second quadrupole in a quadrupole instrument, it is called tandem MS (MS/MS). MS/MS can sometimes be used to quantitate low levels of target compounds in the presence of a high sample matrix background.
The first quadrupole (Q1) is connected with a collision cell (Q2) and another quadrupole (Q3). Both quadrupoles can be used in scanning or static mode, depending on the type of MS/MS analysis being performed. Types of analysis include product ion scan, precursor ion scan, selected reaction monitoring (SRM) (sometimes referred to as multiple reaction monitoring (MRM)) and neutral loss scan. For example: When Q1 is in static mode (looking at one mass only as in SIM), and Q3 is in scanning mode, one obtains a so-called product ion spectrum (also called «daughter spectrum»). From this spectrum, one can select a prominent product ion which can be the product ion for the chosen precursor ion. The pair is called a «transition» and forms the basis for SRM. SRM is highly specific and virtually eliminates matrix background.
Ionization[edit]
After the molecules travel the length of the column, pass through the transfer line and enter into the mass spectrometer they are ionized by various methods with typically only one method being used at any given time. Once the sample is fragmented it will then be detected, usually by an electron multiplier, which essentially turns the ionized mass fragment into an electrical signal that is then detected.
The ionization technique chosen is independent of using full scan or SIM.
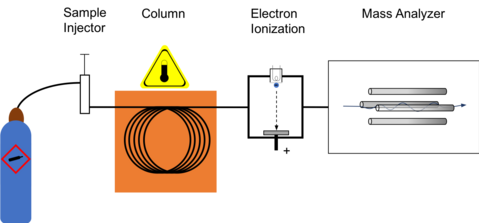
Block diagram for gas chromatography using electron ionization for collecting mass spectrum
Electron ionization[edit]
By far the most common and perhaps standard form of ionization is electron ionization (EI). The molecules enter into the MS (the source is a quadrupole or the ion trap itself in an ion trap MS) where they are bombarded with free electrons emitted from a filament, not unlike the filament one would find in a standard light bulb. The electrons bombard the molecules, causing the molecule to fragment in a characteristic and reproducible way. This «hard ionization» technique results in the creation of more fragments of low mass-to-charge ratio (m/z) and few, if any, molecules approaching the molecular mass unit. Hard ionization is considered by mass spectrometrists as the employ of molecular electron bombardment, whereas «soft ionization» is charge by molecular collision with an introduced gas. The molecular fragmentation pattern is dependent upon the electron energy applied to the system, typically 70 eV (electronvolts). The use of 70 eV facilitates comparison of generated spectra with library spectra using manufacturer-supplied software or software developed by the National Institute of Standards (NIST-USA). Spectral library searches employ matching algorithms such as Probability Based Matching[12] and dot-product[13] matching that are used with methods of analysis written by many method standardization agencies. Sources of libraries include NIST,[14] Wiley,[15] the AAFS,[16] and instrument manufacturers.
Cold electron ionization[edit]
The «hard ionization» process of electron ionization can be softened by the cooling of the molecules before their ionization, resulting in mass spectra that are richer in information.[17][18] In this method named cold electron ionization (cold-EI) the molecules exit the GC column, mixed with added helium make up gas and expand into vacuum through a specially designed supersonic nozzle, forming a supersonic molecular beam (SMB). Collisions with the make up gas at the expanding supersonic jet reduce the internal vibrational (and rotational) energy of the analyte molecules, hence reducing the degree of fragmentation caused by the electrons during the ionization process.[17][18] Cold-EI mass spectra are characterized by an abundant molecular ion while the usual fragmentation pattern is retained, thus making cold-EI mass spectra compatible with library search identification techniques. The enhanced molecular ions increase the identification probabilities of both known and unknown compounds, amplify isomer mass spectral effects and enable the use of isotope abundance analysis for the elucidation of elemental formulas.[19]
Chemical ionization[edit]
In chemical ionization (CI) a reagent gas, typically methane or ammonia is introduced into the mass spectrometer. Depending on the technique (positive CI or negative CI) chosen, this reagent gas will interact with the electrons and analyte and cause a ‘soft’ ionization of the molecule of interest. A softer ionization fragments the molecule to a lower degree than the hard ionization of EI. One of the main benefits of using chemical ionization is that a mass fragment closely corresponding to the molecular weight of the analyte of interest is produced.
In positive chemical ionization (PCI) the reagent gas interacts with the target molecule, most often with a proton exchange. This produces the species in relatively high amounts.
In negative chemical ionization (NCI) the reagent gas decreases the impact of the free electrons on the target analyte. This decreased energy typically leaves the fragment in great supply.
Analysis[edit]
A mass spectrometer is typically utilized in one of two ways: full scan or selective ion monitoring (SIM). The typical GC-MS instrument is capable of performing both functions either individually or concomitantly, depending on the setup of the particular instrument.
The primary goal of instrument analysis is to quantify an amount of substance. This is done by comparing the relative concentrations among the atomic masses in the generated spectrum. Two kinds of analysis are possible, comparative and original. Comparative analysis essentially compares the given spectrum to a spectrum library to see if its characteristics are present for some sample in the library. This is best performed by a computer because there are a myriad of visual distortions that can take place due to variations in scale. Computers can also simultaneously correlate more data (such as the retention times identified by GC), to more accurately relate certain data. Deep learning was shown to lead to promising results in the identification of VOCs from raw GC-MS data[20]
Another method of analysis measures the peaks in relation to one another. In this method, the tallest peak is assigned 100% of the value, and the other peaks being assigned proportionate values. All values above 3% are assigned. The total mass of the unknown compound is normally indicated by the parent peak. The value of this parent peak can be used to fit with a chemical formula containing the various elements which are believed to be in the compound. The isotope pattern in the spectrum, which is unique for elements that have many natural isotopes, can also be used to identify the various elements present. Once a chemical formula has been matched to the spectrum, the molecular structure and bonding can be identified, and must be consistent with the characteristics recorded by GC-MS. Typically, this identification is done automatically by programs which come with the instrument, given a list of the elements which could be present in the sample.
A “full spectrum” analysis considers all the “peaks” within a spectrum. Conversely, selective ion monitoring (SIM) only monitors selected ions associated with a specific substance. This is done on the assumption that at a given retention time, a set of ions is characteristic of a certain compound. This is a fast and efficient analysis, especially if the analyst has previous information about a sample or is only looking for a few specific substances. When the amount of information collected about the ions in a given gas chromatographic peak decreases, the sensitivity of the analysis increases. So, SIM analysis allows for a smaller quantity of a compound to be detected and measured, but the degree of certainty about the identity of that compound is reduced.
Full scan MS[edit]
When collecting data in the full scan mode, a target range of mass fragments is determined and put into the instrument’s method. An example of a typical broad range of mass fragments to monitor would be m/z 50 to m/z 400. The determination of what range to use is largely dictated by what one anticipates being in the sample while being cognizant of the solvent and other possible interferences. A MS should not be set to look for mass fragments too low or else one may detect air (found as m/z 28 due to nitrogen), carbon dioxide (m/z 44) or other possible interference. Additionally if one is to use a large scan range then sensitivity of the instrument is decreased due to performing fewer scans per second since each scan will have to detect a wide range of mass fragments.
Full scan is useful in determining unknown compounds in a sample. It provides more information than SIM when it comes to confirming or resolving compounds in a sample. During instrument method development it may be common to first analyze test solutions in full scan mode to determine the retention time and the mass fragment fingerprint before moving to a SIM instrument method.
Selective ion monitoring[edit]
In selective ion monitoring (SIM) certain ion fragments are entered into the instrument method and only those mass fragments are detected by the mass spectrometer. The advantages of SIM are that the detection limit is lower since the instrument is only looking at a small number of fragments (e.g. three fragments) during each scan. More scans can take place each second. Since only a few mass fragments of interest are being monitored, matrix interferences are typically lower. To additionally confirm the likelihood of a potentially positive result, it is relatively important to be sure that the ion ratios of the various mass fragments are comparable to a known reference standard.
Applications[edit]
Environmental monitoring and cleanup[edit]
GC-MS is becoming the tool of choice for tracking organic pollutants in the environment. The cost of GC-MS equipment has decreased significantly, and the reliability has increased at the same time, which has contributed to its increased adoption in environmental studies.
Criminal forensics[edit]
GC-MS can analyze the particles from a human body in order to help link a criminal to a crime. The analysis of fire debris using GC-MS is well established, and there is even an established American Society for Testing and Materials (ASTM) standard for fire debris analysis. GCMS/MS is especially useful here as samples often contain very complex matrices and results, used in court, need to be highly accurate.
Law enforcement[edit]
GC-MS is increasingly used for detection of illegal narcotics, and may eventually supplant drug-sniffing dogs.[1] A simple and selective GC-MS method for detecting marijuana usage was recently developed by the Robert Koch-Institute in Germany. It involves identifying an acid metabolite of tetrahydrocannabinol (THC), the active ingredient in marijuana, in urine samples by employing derivatization in the sample preparation.[21] GC-MS is also commonly used in forensic toxicology to find drugs and/or poisons in biological specimens of suspects, victims, or the deceased. In drug screening, GC-MS methods frequently utilize liquid-liquid extraction as a part of sample preparation, in which target compounds are extracted from blood plasma.[22]
Sports anti-doping analysis[edit]
GC-MS is the main tool used in sports anti-doping laboratories to test athletes’ urine samples for prohibited performance-enhancing drugs, for example anabolic steroids.[23]
Security[edit]
A post–September 11 development, explosive detection systems have become a part of all US airports. These systems run on a host of technologies, many of them based on GC-MS. There are only three manufacturers certified by the FAA to provide these systems,[citation needed] one of which is Thermo Detection (formerly Thermedics), which produces the EGIS, a GC-MS-based line of explosives detectors. The other two manufacturers are Barringer Technologies, now owned by Smith ‘s Detection Systems, and Ion Track Instruments, part of General Electric Infrastructure Security Systems.
Chemical warfare agent detection[edit]
As part of the post-September 11 drive towards increased capability in homeland security and public health preparedness, traditional GC-MS units with transmission quadrupole mass spectrometers, as well as those with cylindrical ion trap (CIT-MS) and toroidal ion trap (T-ITMS) mass spectrometers have been modified for field portability and near real-time detection of chemical warfare agents (CWA) such as sarin, soman, and VX.[24] These complex and large GC-MS systems have been modified and configured with resistively heated low thermal mass (LTM) gas chromatographs that reduce analysis time to less than ten percent of the time required in traditional laboratory systems.[25] Additionally, the systems are smaller, and more mobile, including units that are mounted in mobile analytical laboratories (MAL), such as those used by the United States Marine Corps Chemical and Biological Incident Response Force MAL and other similar laboratories, and systems that are hand-carried by two-person teams or individuals, much ado to the smaller mass detectors.[26] Depending on the system, the analytes can be introduced via liquid injection, desorbed from sorbent tubes through a thermal desorption process, or with solid-phase micro extraction (SPME).
Chemical engineering[edit]
GC-MS is used for the analysis of unknown organic compound mixtures. One critical use of this technology is the use of GC-MS to determine the composition of bio-oils processed from raw biomass.[27] GC-MS is also utilized in the identification of continuous phase component in a smart material, Magnetorheological (MR) fluid.[28]
Food, beverage and perfume analysis[edit]
Foods and beverages contain numerous aromatic compounds, some naturally present in the raw materials and some forming during processing. GC-MS is extensively used for the analysis of these compounds which include esters, fatty acids, alcohols, aldehydes, terpenes etc. It is also used to detect and measure contaminants from spoilage or adulteration which may be harmful and which is often controlled by governmental agencies, for example pesticides.
Astrochemistry[edit]
Several GC-MS have left earth. Two were brought to Mars by the Viking program.[29] Venera 11 and 12 and Pioneer Venus analysed the atmosphere of Venus with GC-MS.[30] The Huygens probe of the Cassini–Huygens mission landed one GC-MS on Saturn’s largest moon, Titan.[31] The MSL Curiosity rover’s Sample analysis at Mars (SAM) instrument contains both a gas chromatograph and quadrupole mass spectrometer that can be used in tandem as a GC-MS.[32] The material in the comet 67P/Churyumov–Gerasimenko was analysed by the Rosetta mission with a chiral GC-MS in 2014.[33]
Medicine[edit]
Dozens of congenital metabolic diseases also known as inborn errors of metabolism (IEM) are now detectable by newborn screening tests, especially the testing using gas chromatography–mass spectrometry. GC-MS can determine compounds in urine even in minor concentration. These compounds are normally not present but appear in individuals suffering with metabolic disorders. This is increasingly becoming a common way to diagnose IEM for earlier diagnosis and institution of treatment eventually leading to a better outcome. It is now possible to test a newborn for over 100 genetic metabolic disorders by a urine test at birth based on GC-MS.
In combination with isotopic labeling of metabolic compounds, the GC-MS is used for determining metabolic activity. Most applications are based on the use of 13C as the labeling and the measurement of 13C-12C ratios with an isotope ratio mass spectrometer (IRMS); an MS with a detector designed to measure a few select ions and return values as ratios.
See also[edit]
- Capillary electrophoresis–mass spectrometry
- Ion-mobility spectrometry–mass spectrometry
- Liquid chromatography–mass spectrometry
- Prolate trochoidal mass spectrometer
- Pyrolysis–gas chromatography–mass spectrometry
References[edit]
- ^ Sparkman DO, Penton Z, Kitson FG (17 May 2011). Gas Chromatography and Mass Spectrometry: A Practical Guide. Academic Press. ISBN 978-0-08-092015-3.
- ^ Jones M. «Gas Chromatography-Mass Spectrometry». American Chemical Society. Retrieved 19 Nov 2019.
- ^ Fang M, Ivanisevic J, Benton HP, Johnson CH, Patti GJ, Hoang LT, et al. (November 2015). «Thermal Degradation of Small Molecules: A Global Metabolomic Investigation». Analytical Chemistry. 87 (21): 10935–41. doi:10.1021/acs.analchem.5b03003. PMC 4633772. PMID 26434689.
- ^ Holmes JC, Morrell FA (1957). «Oscillographic Mass Spectrometric Monitoring of Gas Chromatography». Applied Spectroscopy. 11 (2): 86–87. doi:10.1366/000370257774633394. ISSN 0003-7028.
- ^ Gohlke RS (1959). «Time-of-Flight Mass Spectrometry and Gas-Liquid Partition Chromatography». Analytical Chemistry. 31 (4): 535–541. doi:10.1021/ac50164a024. ISSN 0003-2700.
- ^ Patton HW, Lewis JS, Kaye WI (1955). «Separation and Analysis of Gases and Volatile Liquids by Gas Chromatography». Analytical Chemistry. 27 (2): 170–174. doi:10.1021/ac60098a002.
- ^ a b c Brock DC (2011). «A Measure of Success». Chemical Heritage Magazine. 29 (1). Retrieved 22 March 2018.
- ^ Webb-Halpern L (2008). «Detecting Success». Chemical Heritage Magazine. 26 (2): 31.
- ^ «Thermo Instrument Systems Inc. History». International Directory of Company Histories (Volume 11 ed.). St. James Press. 1995. pp. 513–514. Retrieved 23 January 2015.
- ^ «Optimizing the Analysis of Volatile Organic Compounds – Technical Guide» Restek Corporation, Lit. Cat. 59887A
- ^ Wang T, Lenahan R (April 1984). «Determination of volatile halocarbons in water by purge-closed loop gas chromatography». Bulletin of Environmental Contamination and Toxicology. 32 (4): 429–38. doi:10.1007/BF01607519. PMID 6713137. S2CID 992748.
- ^ Stauffer DB, McLafferty FW, Ellis RD, Peterson DW (1974). «Probability based matching of mass spectra. Rapid identification of specific compounds in mixtures». Organic Mass Spectrometry. 9 (4): 690–702. doi:10.1002/oms.1210090710.
- ^ Stein SE, Scott DR (September 1994). «Optimization and testing of mass spectral library search algorithms for compound identification». Journal of the American Society for Mass Spectrometry. 5 (9): 859–66. doi:10.1016/1044-0305(94)87009-8. PMID 24222034.
- ^ Standard Reference Data. nist.gov
- ^ Wiley’s Scientific, Technical, and Medical Databases: Home. wiley.com
- ^ Mass Spectrometry Database Committee. ualberta.ca
- ^ a b Amirav A, Gordin A, Poliak M, Fialkov AB (February 2008). «Gas chromatography-mass spectrometry with supersonic molecular beams». Journal of Mass Spectrometry. 43 (2): 141–63. Bibcode:2008JMSp…43..141A. doi:10.1002/jms.1380. PMID 18225851.
- ^ a b SMB-MS (Supersonic GC-MS). tau.ac.il
- ^ Alon T, Amirav A (2006). «Isotope abundance analysis methods and software for improved sample identification with supersonic gas chromatography/mass spectrometry». Rapid Communications in Mass Spectrometry. 20 (17): 2579–88. Bibcode:2006RCMS…20.2579A. doi:10.1002/rcm.2637. PMID 16897787.
- ^ Skarysz A (July 2018). «Convolutional neural networks for automated targeted analysis of raw gas chromatography-mass spectrometry data». International Joint Conferences on Neural Networks (2018) Rio de Janeiro, Brazil: 1–8. doi:10.1109/IJCNN.2018.8489539. ISBN 978-1-5090-6014-6. S2CID 52989098.
- ^ Hübschmann HJ (22 April 2015). Handbook of GC-MS : Fundamentals and Applications (3 ed.). John Wiley & Sons, Incorporated. p. 735. ISBN 9783527674336. Retrieved 22 January 2018.
- ^ Hübschmann HJ (22 April 2015). Handbook of GC-MS : Fundamentals and Applications (3 ed.). John Wiley & Sons, Incorporated. p. 731. ISBN 9783527674336. Retrieved 22 January 2018.
- ^ Tsivou M, Kioukia-Fougia N, Lyris E, Aggelis Y, Fragkaki A, Kiousi X, et al. (2006). «An overview of the doping control analysis during the Olympic Games of 2004 in Athens, Greece». Analytica Chimica Acta. 555: 1–13. doi:10.1016/j.aca.2005.08.068.
- ^ Smith PA, Lepage CJ, Lukacs M, Martin N, Shufutinsky A, Savage PB (2010). «Field-portable gas chromatography with transmission quadrupole and cylindrical ion trap mass spectrometric detection: Chromatographic retention index data and ion/molecule interactions for chemical warfare agent identification». International Journal of Mass Spectrometry. 295 (3): 113–118. Bibcode:2010IJMSp.295..113S. doi:10.1016/j.ijms.2010.03.001.
- ^ Sloan KM, Mustacich RV, Eckenrode BA (2001). «Development and evaluation of a low thermal mass gas chromatograph for rapid forensic GC-MS analyses». Field Analytical Chemistry & Technology. 5 (6): 288–301. doi:10.1002/fact.10011.
- ^ Patterson GE, Guymon AJ, Riter LS, Everly M, Griep-Raming J, Laughlin BC, et al. (December 2002). «Miniature cylindrical ion trap mass spectrometer». Analytical Chemistry. 74 (24): 6145–53. doi:10.1021/ac020494d. PMID 12510732.
- ^ Tekin K, Karagöz S, Bektaş S (2014-12-01). «A review of hydrothermal biomass processing». Renewable and Sustainable Energy Reviews. 40: 673–687. doi:10.1016/j.rser.2014.07.216.
- ^ Unuh MH, Muhamad P, Waziralilah NF, Amran MH (2019). «Characterization of Vehicle Smart Fluid using Gas Chromatography-Mass Spectrometry (GCMS)» (PDF). Journal of Advanced Research in Fluid Mechanics and Thermal Sciences. 55 (2): 240–248.
- ^ SEARCHING FOR LIFE ON MARS: The Development of the Viking GCMS. NASA
- ^ Krasnopolsky VA, Parshev VA (1981). «Chemical composition of the atmosphere of Venus». Nature. 292 (5824): 610–613. Bibcode:1981Natur.292..610K. doi:10.1038/292610a0. S2CID 4369293.
- ^ Niemann HB, Atreya SK, Bauer SJ, Carignan GR, Demick JE, Frost RL, et al. (December 2005). «The abundances of constituents of Titan’s atmosphere from the GCMS instrument on the Huygens probe» (PDF). Nature. 438 (7069): 779–84. Bibcode:2005Natur.438..779N. doi:10.1038/nature04122. hdl:2027.42/62703. PMID 16319830. S2CID 4344046.
- ^ «MSL Science Corner: Sample Analysis at Mars (SAM)». msl-scicorner.jpl.nasa.gov. Archived from the original on 2009-03-20. Retrieved 2019-06-25.
- ^ Gösmann F, Rosenbauer H, Roll R, Böhnhardt H (October 2005). «COSAC onboard Rosetta: a bioastronomy experiment for the short-period comet 67P/Churyumov-Gerasimenko». Astrobiology. 5 (5): 622–31. Bibcode:2005AsBio…5..622G. doi:10.1089/ast.2005.5.622. PMID 16225435.
Bibliography[edit]
- Adams RP (2007). Identification of Essential Oil Components By Gas Chromatography/Mass Spectrometry. Allured Pub Corp. ISBN 978-1-932633-21-4.
- Adlard ER, Handley AJ (2001). Gas chromatographic techniques and applications. London: Sheffield Academic. ISBN 978-0-8493-0521-4.
- Barry EF, Grob RE (2004). Modern practice of gas chromatography. New York: Wiley-Interscience. ISBN 978-0-471-22983-4.
- Eiceman GA (2000). «Gas Chromatography». In Meyers RA (ed.). Encyclopedia of Analytical Chemistry: Applications, Theory, and Instrumentation. Chichester: Wiley. p. 10627. ISBN 0-471-97670-9.
- Giannelli PC, Imwinkelried EJ (1999). «Drug Identification: Gas Chromatography.». Scientific Evidence. Vol. 2. Charlottesville: Lexis Law Publishing. p. 362. ISBN 0-327-04985-5.
- McEwen CN, Kitson FG, Larsen BS (1996). Gas chromatography and mass spectrometry: a practical guide. Boston: Academic Press. ISBN 978-0-12-483385-2.
- McMaster C, McMaster MC (1998). GC/MS: a practical user’s guide. New York: Wiley. ISBN 978-0-471-24826-2.
- Message GM (1984). Practical aspects of gas chromatography/mass spectrometry. New York: Wiley. ISBN 978-0-471-06277-6.
- Niessen WM (2001). Current practice of gas chromatography–mass spectrometry. New York, N.Y: Marcel Dekker. ISBN 978-0-8247-0473-5.
- Weber A, Maurer HW, Pfleger K (2007). Mass Spectral and GC Data of Drugs, Poisons, Pesticides, Pollutants and Their Metabolites. Weinheim: Wiley-VCH. ISBN 978-3-527-31538-3.
External links[edit]
- Gas+chromatography-mass+spectrometry at the US National Library of Medicine Medical Subject Headings (MeSH)
- Golm Metabolome Database, a mass spectral reference database of plant metabolites
Example of a GC-MS instrument
Gas chromatography–mass spectrometry (GC-MS) is an analytical method that combines the features of gas-chromatography and mass spectrometry to identify different substances within a test sample.[1] Applications of GC-MS include drug detection, fire investigation, environmental analysis, explosives investigation, and identification of unknown samples, including that of material samples obtained from planet Mars during probe missions as early as the 1970s. GC-MS can also be used in airport security to detect substances in luggage or on human beings. Additionally, it can identify trace elements in materials that were previously thought to have disintegrated beyond identification. Like liquid chromatography–mass spectrometry, it allows analysis and detection even of tiny amounts of a substance.[2]
GC-MS has been regarded as a «gold standard» for forensic substance identification because it is used to perform a 100% specific test, which positively identifies the presence of a particular substance. A nonspecific test merely indicates that any of several in a category of substances is present. Although a nonspecific test could statistically suggest the identity of the substance, this could lead to false positive identification. However, the high temperatures (300°C) used in the GC-MS injection port (and oven) can result in thermal degradation of injected molecules,[3] thus resulting in the measurement of degradation products instead of the actual molecule(s) of interest.
History[edit]
The first on-line coupling of gas chromatography to a mass spectrometer was reported in the late 1950s.[4][5] An interest in coupling the methods had been suggested as early as December 1954.[6]
The development of affordable and miniaturized computers has helped in the simplification of the use of this instrument, as well as allowed great improvements in the amount of time it takes to analyze a sample. In 1964, Electronic Associates, Inc. (EAI), a leading U.S. supplier of analog computers, began development of a computer controlled quadrupole mass spectrometer under the direction of Robert E. Finnigan.[7] By 1966 Finnigan and collaborator Mike Uthe’s EAI division had sold over 500 quadrupole residual gas-analyzer instruments.[7] In 1967, Finnigan left EAI to form the Finnigan Instrument Corporation along with Roger Sant, T. Z. Chou, Michael Story, Lloyd Friedman, and William Fies.[8] In early 1968, they delivered the first prototype quadrupole GC/MS instruments to Stanford and Purdue University.[7] When Finnigan Instrument Corporation was acquired by Thermo Instrument Systems (later Thermo Fisher Scientific) in 1990, it was considered «the world’s leading manufacturer of mass spectrometers».[9]
Instrumentation[edit]
The insides of the GC-MS, with the column of the gas chromatograph in the oven on the right.
The GC-MS is composed of two major building blocks: the gas chromatograph and the mass spectrometer. The gas chromatograph utilizes a capillary column whose properties regarding molecule separation depend on the column’s dimensions (length, diameter, film thickness) as well as the phase properties (e.g. 5% phenyl polysiloxane). The difference in the chemical properties between different molecules in a mixture and their relative affinity for the stationary phase of the column will promote separation of the molecules as the sample travels the length of the column. The molecules are retained by the column and then elute (come off) from the column at different times (called the retention time), and this allows the mass spectrometer downstream to capture, ionize, accelerate, deflect, and detect the ionized molecules separately. The mass spectrometer does this by breaking each molecule into ionized fragments and detecting these fragments using their mass-to-charge ratio.
These two components, used together, allow a much finer degree of substance identification than either unit used separately. It is not possible to make an accurate identification of a particular molecule by gas chromatography or mass spectrometry alone. The mass spectrometry process normally requires a very pure sample while gas chromatography using a traditional detector (e.g. Flame ionization detector) cannot differentiate between multiple molecules that happen to take the same amount of time to travel through the column (i.e. have the same retention time), which results in two or more molecules that co-elute. Sometimes two different molecules can also have a similar pattern of ionized fragments in a mass spectrometer (mass spectrum). Combining the two processes reduces the possibility of error, as it is extremely unlikely that two different molecules will behave in the same way in both a gas chromatograph and a mass spectrometer. Therefore, when an identifying mass spectrum appears at a characteristic retention time in a GC-MS analysis, it typically increases certainty that the analyte of interest is in the sample.
Purge and trap GC-MS[edit]
For the analysis of volatile compounds, a purge and trap (P&T) concentrator system may be used to introduce samples. The target analytes are extracted by mixing the sample with water and purge with inert gas (e.g. Nitrogen gas) into an airtight chamber, this is known as purging or sparging. The volatile compounds move into the headspace above the water and are drawn along a pressure gradient (caused by the introduction of the purge gas) out of the chamber. The volatile compounds are drawn along a heated line onto a ‘trap’. The trap is a column of adsorbent material at ambient temperature that holds the compounds by returning them to the liquid phase. The trap is then heated and the sample compounds are introduced to the GC-MS column via a volatiles interface, which is a split inlet system. P&T GC-MS is particularly suited to volatile organic compounds (VOCs) and BTEX compounds (aromatic compounds associated with petroleum).[10]
A faster alternative is the «purge-closed loop» system. In this system the inert gas is bubbled through the water until the concentrations of organic compounds in the vapor phase are at equilibrium with concentrations in the aqueous phase. The gas phase is then analysed directly.[11]
Types of mass spectrometer detectors[edit]
The most common type of mass spectrometer (MS) associated with a gas chromatograph (GC) is the quadrupole mass spectrometer, sometimes referred to by the Hewlett-Packard (now Agilent) trade name «Mass Selective Detector» (MSD). Another relatively common detector is the ion trap mass spectrometer. Additionally one may find a magnetic sector mass spectrometer, however these particular instruments are expensive and bulky and not typically found in high-throughput service laboratories. Other detectors may be encountered such as time of flight (TOF), tandem quadrupoles (MS-MS) (see below), or in the case of an ion trap MSn where n indicates the number mass spectrometry stages.
GC-tandem MS[edit]
When a second phase of mass fragmentation is added, for example using a second quadrupole in a quadrupole instrument, it is called tandem MS (MS/MS). MS/MS can sometimes be used to quantitate low levels of target compounds in the presence of a high sample matrix background.
The first quadrupole (Q1) is connected with a collision cell (Q2) and another quadrupole (Q3). Both quadrupoles can be used in scanning or static mode, depending on the type of MS/MS analysis being performed. Types of analysis include product ion scan, precursor ion scan, selected reaction monitoring (SRM) (sometimes referred to as multiple reaction monitoring (MRM)) and neutral loss scan. For example: When Q1 is in static mode (looking at one mass only as in SIM), and Q3 is in scanning mode, one obtains a so-called product ion spectrum (also called «daughter spectrum»). From this spectrum, one can select a prominent product ion which can be the product ion for the chosen precursor ion. The pair is called a «transition» and forms the basis for SRM. SRM is highly specific and virtually eliminates matrix background.
Ionization[edit]
After the molecules travel the length of the column, pass through the transfer line and enter into the mass spectrometer they are ionized by various methods with typically only one method being used at any given time. Once the sample is fragmented it will then be detected, usually by an electron multiplier, which essentially turns the ionized mass fragment into an electrical signal that is then detected.
The ionization technique chosen is independent of using full scan or SIM.
Block diagram for gas chromatography using electron ionization for collecting mass spectrum
Electron ionization[edit]
By far the most common and perhaps standard form of ionization is electron ionization (EI). The molecules enter into the MS (the source is a quadrupole or the ion trap itself in an ion trap MS) where they are bombarded with free electrons emitted from a filament, not unlike the filament one would find in a standard light bulb. The electrons bombard the molecules, causing the molecule to fragment in a characteristic and reproducible way. This «hard ionization» technique results in the creation of more fragments of low mass-to-charge ratio (m/z) and few, if any, molecules approaching the molecular mass unit. Hard ionization is considered by mass spectrometrists as the employ of molecular electron bombardment, whereas «soft ionization» is charge by molecular collision with an introduced gas. The molecular fragmentation pattern is dependent upon the electron energy applied to the system, typically 70 eV (electronvolts). The use of 70 eV facilitates comparison of generated spectra with library spectra using manufacturer-supplied software or software developed by the National Institute of Standards (NIST-USA). Spectral library searches employ matching algorithms such as Probability Based Matching[12] and dot-product[13] matching that are used with methods of analysis written by many method standardization agencies. Sources of libraries include NIST,[14] Wiley,[15] the AAFS,[16] and instrument manufacturers.
Cold electron ionization[edit]
The «hard ionization» process of electron ionization can be softened by the cooling of the molecules before their ionization, resulting in mass spectra that are richer in information.[17][18] In this method named cold electron ionization (cold-EI) the molecules exit the GC column, mixed with added helium make up gas and expand into vacuum through a specially designed supersonic nozzle, forming a supersonic molecular beam (SMB). Collisions with the make up gas at the expanding supersonic jet reduce the internal vibrational (and rotational) energy of the analyte molecules, hence reducing the degree of fragmentation caused by the electrons during the ionization process.[17][18] Cold-EI mass spectra are characterized by an abundant molecular ion while the usual fragmentation pattern is retained, thus making cold-EI mass spectra compatible with library search identification techniques. The enhanced molecular ions increase the identification probabilities of both known and unknown compounds, amplify isomer mass spectral effects and enable the use of isotope abundance analysis for the elucidation of elemental formulas.[19]
Chemical ionization[edit]
In chemical ionization (CI) a reagent gas, typically methane or ammonia is introduced into the mass spectrometer. Depending on the technique (positive CI or negative CI) chosen, this reagent gas will interact with the electrons and analyte and cause a ‘soft’ ionization of the molecule of interest. A softer ionization fragments the molecule to a lower degree than the hard ionization of EI. One of the main benefits of using chemical ionization is that a mass fragment closely corresponding to the molecular weight of the analyte of interest is produced.
In positive chemical ionization (PCI) the reagent gas interacts with the target molecule, most often with a proton exchange. This produces the species in relatively high amounts.
In negative chemical ionization (NCI) the reagent gas decreases the impact of the free electrons on the target analyte. This decreased energy typically leaves the fragment in great supply.
Analysis[edit]
A mass spectrometer is typically utilized in one of two ways: full scan or selective ion monitoring (SIM). The typical GC-MS instrument is capable of performing both functions either individually or concomitantly, depending on the setup of the particular instrument.
The primary goal of instrument analysis is to quantify an amount of substance. This is done by comparing the relative concentrations among the atomic masses in the generated spectrum. Two kinds of analysis are possible, comparative and original. Comparative analysis essentially compares the given spectrum to a spectrum library to see if its characteristics are present for some sample in the library. This is best performed by a computer because there are a myriad of visual distortions that can take place due to variations in scale. Computers can also simultaneously correlate more data (such as the retention times identified by GC), to more accurately relate certain data. Deep learning was shown to lead to promising results in the identification of VOCs from raw GC-MS data[20]
Another method of analysis measures the peaks in relation to one another. In this method, the tallest peak is assigned 100% of the value, and the other peaks being assigned proportionate values. All values above 3% are assigned. The total mass of the unknown compound is normally indicated by the parent peak. The value of this parent peak can be used to fit with a chemical formula containing the various elements which are believed to be in the compound. The isotope pattern in the spectrum, which is unique for elements that have many natural isotopes, can also be used to identify the various elements present. Once a chemical formula has been matched to the spectrum, the molecular structure and bonding can be identified, and must be consistent with the characteristics recorded by GC-MS. Typically, this identification is done automatically by programs which come with the instrument, given a list of the elements which could be present in the sample.
A “full spectrum” analysis considers all the “peaks” within a spectrum. Conversely, selective ion monitoring (SIM) only monitors selected ions associated with a specific substance. This is done on the assumption that at a given retention time, a set of ions is characteristic of a certain compound. This is a fast and efficient analysis, especially if the analyst has previous information about a sample or is only looking for a few specific substances. When the amount of information collected about the ions in a given gas chromatographic peak decreases, the sensitivity of the analysis increases. So, SIM analysis allows for a smaller quantity of a compound to be detected and measured, but the degree of certainty about the identity of that compound is reduced.
Full scan MS[edit]
When collecting data in the full scan mode, a target range of mass fragments is determined and put into the instrument’s method. An example of a typical broad range of mass fragments to monitor would be m/z 50 to m/z 400. The determination of what range to use is largely dictated by what one anticipates being in the sample while being cognizant of the solvent and other possible interferences. A MS should not be set to look for mass fragments too low or else one may detect air (found as m/z 28 due to nitrogen), carbon dioxide (m/z 44) or other possible interference. Additionally if one is to use a large scan range then sensitivity of the instrument is decreased due to performing fewer scans per second since each scan will have to detect a wide range of mass fragments.
Full scan is useful in determining unknown compounds in a sample. It provides more information than SIM when it comes to confirming or resolving compounds in a sample. During instrument method development it may be common to first analyze test solutions in full scan mode to determine the retention time and the mass fragment fingerprint before moving to a SIM instrument method.
Selective ion monitoring[edit]
In selective ion monitoring (SIM) certain ion fragments are entered into the instrument method and only those mass fragments are detected by the mass spectrometer. The advantages of SIM are that the detection limit is lower since the instrument is only looking at a small number of fragments (e.g. three fragments) during each scan. More scans can take place each second. Since only a few mass fragments of interest are being monitored, matrix interferences are typically lower. To additionally confirm the likelihood of a potentially positive result, it is relatively important to be sure that the ion ratios of the various mass fragments are comparable to a known reference standard.
Applications[edit]
Environmental monitoring and cleanup[edit]
GC-MS is becoming the tool of choice for tracking organic pollutants in the environment. The cost of GC-MS equipment has decreased significantly, and the reliability has increased at the same time, which has contributed to its increased adoption in environmental studies.
Criminal forensics[edit]
GC-MS can analyze the particles from a human body in order to help link a criminal to a crime. The analysis of fire debris using GC-MS is well established, and there is even an established American Society for Testing and Materials (ASTM) standard for fire debris analysis. GCMS/MS is especially useful here as samples often contain very complex matrices and results, used in court, need to be highly accurate.
Law enforcement[edit]
GC-MS is increasingly used for detection of illegal narcotics, and may eventually supplant drug-sniffing dogs.[1] A simple and selective GC-MS method for detecting marijuana usage was recently developed by the Robert Koch-Institute in Germany. It involves identifying an acid metabolite of tetrahydrocannabinol (THC), the active ingredient in marijuana, in urine samples by employing derivatization in the sample preparation.[21] GC-MS is also commonly used in forensic toxicology to find drugs and/or poisons in biological specimens of suspects, victims, or the deceased. In drug screening, GC-MS methods frequently utilize liquid-liquid extraction as a part of sample preparation, in which target compounds are extracted from blood plasma.[22]
Sports anti-doping analysis[edit]
GC-MS is the main tool used in sports anti-doping laboratories to test athletes’ urine samples for prohibited performance-enhancing drugs, for example anabolic steroids.[23]
Security[edit]
A post–September 11 development, explosive detection systems have become a part of all US airports. These systems run on a host of technologies, many of them based on GC-MS. There are only three manufacturers certified by the FAA to provide these systems,[citation needed] one of which is Thermo Detection (formerly Thermedics), which produces the EGIS, a GC-MS-based line of explosives detectors. The other two manufacturers are Barringer Technologies, now owned by Smith ‘s Detection Systems, and Ion Track Instruments, part of General Electric Infrastructure Security Systems.
Chemical warfare agent detection[edit]
As part of the post-September 11 drive towards increased capability in homeland security and public health preparedness, traditional GC-MS units with transmission quadrupole mass spectrometers, as well as those with cylindrical ion trap (CIT-MS) and toroidal ion trap (T-ITMS) mass spectrometers have been modified for field portability and near real-time detection of chemical warfare agents (CWA) such as sarin, soman, and VX.[24] These complex and large GC-MS systems have been modified and configured with resistively heated low thermal mass (LTM) gas chromatographs that reduce analysis time to less than ten percent of the time required in traditional laboratory systems.[25] Additionally, the systems are smaller, and more mobile, including units that are mounted in mobile analytical laboratories (MAL), such as those used by the United States Marine Corps Chemical and Biological Incident Response Force MAL and other similar laboratories, and systems that are hand-carried by two-person teams or individuals, much ado to the smaller mass detectors.[26] Depending on the system, the analytes can be introduced via liquid injection, desorbed from sorbent tubes through a thermal desorption process, or with solid-phase micro extraction (SPME).
Chemical engineering[edit]
GC-MS is used for the analysis of unknown organic compound mixtures. One critical use of this technology is the use of GC-MS to determine the composition of bio-oils processed from raw biomass.[27] GC-MS is also utilized in the identification of continuous phase component in a smart material, Magnetorheological (MR) fluid.[28]
Food, beverage and perfume analysis[edit]
Foods and beverages contain numerous aromatic compounds, some naturally present in the raw materials and some forming during processing. GC-MS is extensively used for the analysis of these compounds which include esters, fatty acids, alcohols, aldehydes, terpenes etc. It is also used to detect and measure contaminants from spoilage or adulteration which may be harmful and which is often controlled by governmental agencies, for example pesticides.
Astrochemistry[edit]
Several GC-MS have left earth. Two were brought to Mars by the Viking program.[29] Venera 11 and 12 and Pioneer Venus analysed the atmosphere of Venus with GC-MS.[30] The Huygens probe of the Cassini–Huygens mission landed one GC-MS on Saturn’s largest moon, Titan.[31] The MSL Curiosity rover’s Sample analysis at Mars (SAM) instrument contains both a gas chromatograph and quadrupole mass spectrometer that can be used in tandem as a GC-MS.[32] The material in the comet 67P/Churyumov–Gerasimenko was analysed by the Rosetta mission with a chiral GC-MS in 2014.[33]
Medicine[edit]
Dozens of congenital metabolic diseases also known as inborn errors of metabolism (IEM) are now detectable by newborn screening tests, especially the testing using gas chromatography–mass spectrometry. GC-MS can determine compounds in urine even in minor concentration. These compounds are normally not present but appear in individuals suffering with metabolic disorders. This is increasingly becoming a common way to diagnose IEM for earlier diagnosis and institution of treatment eventually leading to a better outcome. It is now possible to test a newborn for over 100 genetic metabolic disorders by a urine test at birth based on GC-MS.
In combination with isotopic labeling of metabolic compounds, the GC-MS is used for determining metabolic activity. Most applications are based on the use of 13C as the labeling and the measurement of 13C-12C ratios with an isotope ratio mass spectrometer (IRMS); an MS with a detector designed to measure a few select ions and return values as ratios.
See also[edit]
- Capillary electrophoresis–mass spectrometry
- Ion-mobility spectrometry–mass spectrometry
- Liquid chromatography–mass spectrometry
- Prolate trochoidal mass spectrometer
- Pyrolysis–gas chromatography–mass spectrometry
References[edit]
- ^ Sparkman DO, Penton Z, Kitson FG (17 May 2011). Gas Chromatography and Mass Spectrometry: A Practical Guide. Academic Press. ISBN 978-0-08-092015-3.
- ^ Jones M. «Gas Chromatography-Mass Spectrometry». American Chemical Society. Retrieved 19 Nov 2019.
- ^ Fang M, Ivanisevic J, Benton HP, Johnson CH, Patti GJ, Hoang LT, et al. (November 2015). «Thermal Degradation of Small Molecules: A Global Metabolomic Investigation». Analytical Chemistry. 87 (21): 10935–41. doi:10.1021/acs.analchem.5b03003. PMC 4633772. PMID 26434689.
- ^ Holmes JC, Morrell FA (1957). «Oscillographic Mass Spectrometric Monitoring of Gas Chromatography». Applied Spectroscopy. 11 (2): 86–87. doi:10.1366/000370257774633394. ISSN 0003-7028.
- ^ Gohlke RS (1959). «Time-of-Flight Mass Spectrometry and Gas-Liquid Partition Chromatography». Analytical Chemistry. 31 (4): 535–541. doi:10.1021/ac50164a024. ISSN 0003-2700.
- ^ Patton HW, Lewis JS, Kaye WI (1955). «Separation and Analysis of Gases and Volatile Liquids by Gas Chromatography». Analytical Chemistry. 27 (2): 170–174. doi:10.1021/ac60098a002.
- ^ a b c Brock DC (2011). «A Measure of Success». Chemical Heritage Magazine. 29 (1). Retrieved 22 March 2018.
- ^ Webb-Halpern L (2008). «Detecting Success». Chemical Heritage Magazine. 26 (2): 31.
- ^ «Thermo Instrument Systems Inc. History». International Directory of Company Histories (Volume 11 ed.). St. James Press. 1995. pp. 513–514. Retrieved 23 January 2015.
- ^ «Optimizing the Analysis of Volatile Organic Compounds – Technical Guide» Restek Corporation, Lit. Cat. 59887A
- ^ Wang T, Lenahan R (April 1984). «Determination of volatile halocarbons in water by purge-closed loop gas chromatography». Bulletin of Environmental Contamination and Toxicology. 32 (4): 429–38. doi:10.1007/BF01607519. PMID 6713137. S2CID 992748.
- ^ Stauffer DB, McLafferty FW, Ellis RD, Peterson DW (1974). «Probability based matching of mass spectra. Rapid identification of specific compounds in mixtures». Organic Mass Spectrometry. 9 (4): 690–702. doi:10.1002/oms.1210090710.
- ^ Stein SE, Scott DR (September 1994). «Optimization and testing of mass spectral library search algorithms for compound identification». Journal of the American Society for Mass Spectrometry. 5 (9): 859–66. doi:10.1016/1044-0305(94)87009-8. PMID 24222034.
- ^ Standard Reference Data. nist.gov
- ^ Wiley’s Scientific, Technical, and Medical Databases: Home. wiley.com
- ^ Mass Spectrometry Database Committee. ualberta.ca
- ^ a b Amirav A, Gordin A, Poliak M, Fialkov AB (February 2008). «Gas chromatography-mass spectrometry with supersonic molecular beams». Journal of Mass Spectrometry. 43 (2): 141–63. Bibcode:2008JMSp…43..141A. doi:10.1002/jms.1380. PMID 18225851.
- ^ a b SMB-MS (Supersonic GC-MS). tau.ac.il
- ^ Alon T, Amirav A (2006). «Isotope abundance analysis methods and software for improved sample identification with supersonic gas chromatography/mass spectrometry». Rapid Communications in Mass Spectrometry. 20 (17): 2579–88. Bibcode:2006RCMS…20.2579A. doi:10.1002/rcm.2637. PMID 16897787.
- ^ Skarysz A (July 2018). «Convolutional neural networks for automated targeted analysis of raw gas chromatography-mass spectrometry data». International Joint Conferences on Neural Networks (2018) Rio de Janeiro, Brazil: 1–8. doi:10.1109/IJCNN.2018.8489539. ISBN 978-1-5090-6014-6. S2CID 52989098.
- ^ Hübschmann HJ (22 April 2015). Handbook of GC-MS : Fundamentals and Applications (3 ed.). John Wiley & Sons, Incorporated. p. 735. ISBN 9783527674336. Retrieved 22 January 2018.
- ^ Hübschmann HJ (22 April 2015). Handbook of GC-MS : Fundamentals and Applications (3 ed.). John Wiley & Sons, Incorporated. p. 731. ISBN 9783527674336. Retrieved 22 January 2018.
- ^ Tsivou M, Kioukia-Fougia N, Lyris E, Aggelis Y, Fragkaki A, Kiousi X, et al. (2006). «An overview of the doping control analysis during the Olympic Games of 2004 in Athens, Greece». Analytica Chimica Acta. 555: 1–13. doi:10.1016/j.aca.2005.08.068.
- ^ Smith PA, Lepage CJ, Lukacs M, Martin N, Shufutinsky A, Savage PB (2010). «Field-portable gas chromatography with transmission quadrupole and cylindrical ion trap mass spectrometric detection: Chromatographic retention index data and ion/molecule interactions for chemical warfare agent identification». International Journal of Mass Spectrometry. 295 (3): 113–118. Bibcode:2010IJMSp.295..113S. doi:10.1016/j.ijms.2010.03.001.
- ^ Sloan KM, Mustacich RV, Eckenrode BA (2001). «Development and evaluation of a low thermal mass gas chromatograph for rapid forensic GC-MS analyses». Field Analytical Chemistry & Technology. 5 (6): 288–301. doi:10.1002/fact.10011.
- ^ Patterson GE, Guymon AJ, Riter LS, Everly M, Griep-Raming J, Laughlin BC, et al. (December 2002). «Miniature cylindrical ion trap mass spectrometer». Analytical Chemistry. 74 (24): 6145–53. doi:10.1021/ac020494d. PMID 12510732.
- ^ Tekin K, Karagöz S, Bektaş S (2014-12-01). «A review of hydrothermal biomass processing». Renewable and Sustainable Energy Reviews. 40: 673–687. doi:10.1016/j.rser.2014.07.216.
- ^ Unuh MH, Muhamad P, Waziralilah NF, Amran MH (2019). «Characterization of Vehicle Smart Fluid using Gas Chromatography-Mass Spectrometry (GCMS)» (PDF). Journal of Advanced Research in Fluid Mechanics and Thermal Sciences. 55 (2): 240–248.
- ^ SEARCHING FOR LIFE ON MARS: The Development of the Viking GCMS. NASA
- ^ Krasnopolsky VA, Parshev VA (1981). «Chemical composition of the atmosphere of Venus». Nature. 292 (5824): 610–613. Bibcode:1981Natur.292..610K. doi:10.1038/292610a0. S2CID 4369293.
- ^ Niemann HB, Atreya SK, Bauer SJ, Carignan GR, Demick JE, Frost RL, et al. (December 2005). «The abundances of constituents of Titan’s atmosphere from the GCMS instrument on the Huygens probe» (PDF). Nature. 438 (7069): 779–84. Bibcode:2005Natur.438..779N. doi:10.1038/nature04122. hdl:2027.42/62703. PMID 16319830. S2CID 4344046.
- ^ «MSL Science Corner: Sample Analysis at Mars (SAM)». msl-scicorner.jpl.nasa.gov. Archived from the original on 2009-03-20. Retrieved 2019-06-25.
- ^ Gösmann F, Rosenbauer H, Roll R, Böhnhardt H (October 2005). «COSAC onboard Rosetta: a bioastronomy experiment for the short-period comet 67P/Churyumov-Gerasimenko». Astrobiology. 5 (5): 622–31. Bibcode:2005AsBio…5..622G. doi:10.1089/ast.2005.5.622. PMID 16225435.
Bibliography[edit]
- Adams RP (2007). Identification of Essential Oil Components By Gas Chromatography/Mass Spectrometry. Allured Pub Corp. ISBN 978-1-932633-21-4.
- Adlard ER, Handley AJ (2001). Gas chromatographic techniques and applications. London: Sheffield Academic. ISBN 978-0-8493-0521-4.
- Barry EF, Grob RE (2004). Modern practice of gas chromatography. New York: Wiley-Interscience. ISBN 978-0-471-22983-4.
- Eiceman GA (2000). «Gas Chromatography». In Meyers RA (ed.). Encyclopedia of Analytical Chemistry: Applications, Theory, and Instrumentation. Chichester: Wiley. p. 10627. ISBN 0-471-97670-9.
- Giannelli PC, Imwinkelried EJ (1999). «Drug Identification: Gas Chromatography.». Scientific Evidence. Vol. 2. Charlottesville: Lexis Law Publishing. p. 362. ISBN 0-327-04985-5.
- McEwen CN, Kitson FG, Larsen BS (1996). Gas chromatography and mass spectrometry: a practical guide. Boston: Academic Press. ISBN 978-0-12-483385-2.
- McMaster C, McMaster MC (1998). GC/MS: a practical user’s guide. New York: Wiley. ISBN 978-0-471-24826-2.
- Message GM (1984). Practical aspects of gas chromatography/mass spectrometry. New York: Wiley. ISBN 978-0-471-06277-6.
- Niessen WM (2001). Current practice of gas chromatography–mass spectrometry. New York, N.Y: Marcel Dekker. ISBN 978-0-8247-0473-5.
- Weber A, Maurer HW, Pfleger K (2007). Mass Spectral and GC Data of Drugs, Poisons, Pesticides, Pollutants and Their Metabolites. Weinheim: Wiley-VCH. ISBN 978-3-527-31538-3.
External links[edit]
- Gas+chromatography-mass+spectrometry at the US National Library of Medicine Medical Subject Headings (MeSH)
- Golm Metabolome Database, a mass spectral reference database of plant metabolites
|
Ion trap LCMS system with ESI interface |
|
| Acronym | LCMS |
|---|---|
| Classification | Chromatography Mass spectrometry |
| Analytes | organic molecules biomolecules |
| Manufacturers | Agilent Bruker PerkinElmer SCIEX Shimadzu Scientific Thermo Fisher Scientific Waters Corporation |
| Other techniques | |
| Related | Gas chromatography–mass spectrometry |

Liquid chromatography–mass spectrometry (LC–MS) is an analytical chemistry technique that combines the physical separation capabilities of liquid chromatography (or HPLC) with the mass analysis capabilities of mass spectrometry (MS). Coupled chromatography — MS systems are popular in chemical analysis because the individual capabilities of each technique are enhanced synergistically. While liquid chromatography separates mixtures with multiple components, mass spectrometry provides spectral information that may help to identify (or confirm the suspected identity of) each separated component.[1] MS is not only sensitive, but provides selective detection, relieving the need for complete chromatographic separation.[2] LC-MS is also appropriate for metabolomics because of its good coverage of a wide range of chemicals.[3] This tandem technique can be used to analyze biochemical, organic, and inorganic compounds commonly found in complex samples of environmental and biological origin. Therefore, LC-MS may be applied in a wide range of sectors including biotechnology, environment monitoring, food processing, and pharmaceutical, agrochemical, and cosmetic industries.[4][5] Since the early 2000s, LC-MS (or more specifically LC-MS-MS) has also begun to be used in clinical applications.[6]
In addition to the liquid chromatography and mass spectrometry devices, an LC-MS system contains an interface that efficiently transfers the separated components from the LC column into the MS ion source.[5][7] The interface is necessary because the LC and MS devices are fundamentally incompatible. While the mobile phase in a LC system is a pressurized liquid, the MS analyzers commonly operate under high vacuum. Thus, it is not possible to directly pump the eluate from the LC column into the MS source. Overall, the interface is a mechanically simple part of the LC-MS system that transfers the maximum amount of analyte, removes a significant portion of the mobile phase used in LC and preserves the chemical identity of the chromatography products (chemically inert). As a requirement, the interface should not interfere with the ionizing efficiency and vacuum conditions of the MS system.[5] Nowadays, most extensively applied LC-MS interfaces are based on atmospheric pressure ionization (API) strategies like electrospray ionization (ESI), atmospheric-pressure chemical ionization (APCI), and atmospheric pressure photoionization (APPI). These interfaces became available in the 1990s after a two decade long research and development process.[8][7]
History of LC-MS[edit]
The coupling of chromatography with MS is a well developed chemical analysis strategy dating back from the 1950s. Gas chromatography (GC)–MS was originally introduced in 1952, when A. T. James and A. J. P. Martin were trying to develop tandem separation — mass analysis techniques.[9] In GC, the analytes are eluted from the separation column as a gas and the connection with electron ionization (EI) or chemical ionization (CI) ion sources in the MS system was a technically simpler challenge. Because of this, the development of GC-MS systems was faster than LC-MS and such systems were first commercialized in the 1970s.[7] The development of LC-MS systems took longer than GC-MS and was directly related to the development of proper interfaces. V. L. Tal’roze and collaborators started the development of LC-MS in the late 1960s,[10][11] when they first used capillaries to connect an LC columns to an EI source.[12][8] A similar strategy was investigated by McLafferty and collaborators in 1973 who coupled the LC column to a CI source,[13] which allowed a higher liquid flow into the source. This was the first and most obvious way of coupling LC with MS, and was known as the capillary inlet interface. This pioneer interface for LC-MS had the same analysis capabilities of GC-MS and was limited to rather volatile analytes and non-polar compounds with low molecular mass (below 400 Da). In the capillary inlet interface, the evaporation of the mobile phase inside the capillary was one of the main issues. Within the first years of development of LC-MS, on-line and off-line alternatives were proposed as coupling alternatives. In general, off-line coupling involved fraction collection, evaporation of solvent, and transfer of analytes to the MS using probes. Off-line analyte treatment process was time consuming and there was an inherent risk of sample contamination. Rapidly, it was realized that the analysis of complex mixtures would require the development of a fully automated on-line coupling solution in LC-MS.[8]
The key to the success and wide-spread adoption of LC-MS as a routine analytical tool lies in the interface and ion source between the liquid-based LC and the vacuum-base MS. The following interfaces were stepping-stones on the way to the modern atmospheric-pressure ionization interfaces, and are described for historical interest.
Moving Belt Interface[edit]
The moving-belt interface (MBI) was developed by McFadden et al in 1977 and commercialized by Finnigan.[14] This interface consisted of an endless moving belt onto which the LC column effluent was deposited in a band. On the belt, the solvent was evaporated by gently heating and efficiently exhausting the solvent vapours under reduced pressure in two vacuum chambers. After the liquid phase was removed, the belt passed over a heater which flash desorbed the analytes into the MS ion source. One of the significant advantages of the MBI was its compatibility with a wide range of chromatographic conditions.[8] MBI was successfully used for LC-MS applications between 1978 and 1990 because it allowed coupling of LC to MS devices using EI, CI, and fast-atom bombardment (FAB) ion sources. The most common MS systems connected by MBI interfaces to LC columns were magnetic sector and quadrupole instruments. MBI interfaces for LC-MS allowed MS to be widely applied in the analysis of drugs, pesticides, steroids, alkaloids, and polycyclic aromatic hydrocarbons. This interface is no longer used because of its mechanical complexity and the difficulties associated with belt renewal as well as its inability to handle very labile biomolecules.
Direct liquid introduction interface[edit]
The direct liquid introduction (DLI) interface was developed in 1980. This interface was intended to solve the problem of evaporation of liquid inside the capillary inlet interface. In DLI, a small portion of the LC flow was forced through a small aperture or diaphragm (typically 10um in diameter) to form a liquid jet composed of small droplets that were subsequently dried in a desolvation chamber.[11] The analytes were ionized using a solvent assisted chemical ionization source, where the LC solvents acted as reagent gases. To use this interface, it was necessary to split the flow coming out of the LC column because only a small portion of the effluent (10 to 50 μl/min out of 1 ml/min) could be introduced into the source without raising the vacuum pressure of the MS system too high. Alternately, Henion at Cornell University had success with using micro-bore LC methods so that the entire (low) flow of the LC could be used. One of the main operational problems of the DLI interface was the frequent clogging of the diaphragm orifices. The DLI interface was used between 1982 and 1985 for the analysis of pesticides, corticosteroids, metabolites in horse urine, erythromycin, and vitamin B12. However, this interface was replaced by the thermospray interface, which removed the flow rate limitations and the issues with the clogging diaphragms.[5][8]
A related device was the particle beam interface (PBI), developed by Willoughby and Browner in 1984.[15] Particle beam interfaces took over the wide applications of MBI for LC-MS in 1988.[8][16] The PBI operated by using a helium gas nebulizer to spray the eluant into the vacuum, drying the droplets and pumping away the solvent vapour (using a jet separator) while the stream of monodisperse dried particles containing the analyte entered the source.[11] Drying the droplets outside of the source volume, and using a jet separator to pump away the solvent vapour, allowed the particles to enter and be vapourized in a low-pressure EI source. As with the MBI, the ability to generate library-searchable EI spectra was a distinct advantage for many applications. Commercialized by Hewlett Packard, and later by VG and Extrel, it enjoyed moderate success, but has been largely supplanted by the atmospheric pressure interfaces such as electrospray and APCI which provide a broader range of compound coverage and applications.
Thermospray interface[edit]
The thermospray (TSP) interface was developed in 1980 by Marvin Vestal and co-workers at the University of Houston.[17] It was commercialized by Vestec and several of the major mass spectrometer manufacurers. The interface resulted from a long term research project intended to find a LC-MS interface capable of handling high flow rates (1 ml/min) and avoiding the flow split in DLI interfaces. The TSP interface was composed of a heated probe, a desolvation chamber, and an ion focusing skimmer. The LC effluent passed through the heated probe and emerged as a jet of vapor and small droplets flowing into the desolvation chamber at low pressure. Initially operated with a filament or discharge as the source of ions (thereby acting as a CI source for vapourized analyte), it was soon discovered that ions were also observed when the filament or discharge was off. This could be attributed to either direct emission of ions from the liquid droplets as they evaporated in a process related to electrospray ionization or ion evaporation, or to chemical ionization of vapourized analyte molecules from buffer ions (such as ammonium acetate). The fact that multiply-charged ions were observed from some larger analytes suggests that direct analyte ion emission was occurring under at least some conditions.[11] The interface was able to handle up to 2 ml/min of eluate from the LC column and would efficiently introduce it into the MS vacuum system. TSP was also more suitable for LC-MS applications involving reversed phase liquid chromatography (RT-LC). With time, the mechanical complexity of TSP was simplified, and this interface became popular as the first ideal LC-MS interface for pharmaceutical applications comprising the analysis of drugs, metabolites, conjugates, nucleosides, peptides, natural products, and pesticides. The introduction of TSP marked a significant improvement for LC-MS systems and was the most widely applied interface until the beginning of the 1990s, when it began to be replaced by interfaces involving atmospheric pressure ionization (API).[5][7][16]
FAB based interfaces[edit]
The frit fast atom bombardment (FAB) and continuous flow-FAB (CF-FAB) interfaces were developed in 1985 and 1986 respectively.[16] Both interfaces were similar, but they differed in that the first used a porous frit probe as connecting channel, while CF-FAB used a probe tip. From these, the CF-FAB was more successful as a LC-MS interface and was useful to analyze non-volatile and thermally labile compounds. In these interfaces, the LC effluent passed through the frit or CF-FAB channels to form a uniform liquid film at the tip. There, the liquid was bombarded with ion beams or high energy atoms (fast atoms). For stable operation, the FAB based interfaces were able to handle liquid flow rates of only 1–15 μl and were also restricted to microbore and capillary columns. In order to be used in FAB MS ionization sources, the analytes of interest had to be mixed with a matrix (e.g., glycerol) that could be added before or after the separation in the LC column. FAB based interfaces were extensively used to characterize peptides, but lost applicability with the advent of electrospray based interfaces in 1988.[5][8]
Liquid chromatography[edit]
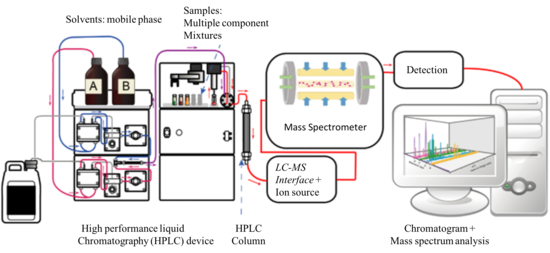
Diagram of an LC-MS system
Liquid chromatography is a method of physical separation in which the components of a liquid mixture are distributed between two immiscible phases, i.e., stationary and mobile. The practice of LC can be divided into five categories, i.e., adsorption chromatography, partition chromatography, ion-exchange chromatography, size-exclusion chromatography, and affinity chromatography. Among these, the most widely used variant is the reverse-phase (RP) mode of the partition chromatography technique, which makes use of a nonpolar (hydrophobic) stationary phase and a polar mobile phase. In common applications, the mobile phase is a mixture of water and other polar solvents (e.g., methanol, isopropanol, and acetonitrile), and the stationary matrix is prepared by attaching long-chain alkyl groups (e.g., n-octadecyl or C18) to the external and internal surfaces of irregularly or spherically shaped 5 μm diameter porous silica particles.[5]
In HPLC, typically 20 μl of the sample of interest are injected into the mobile phase stream delivered by a high pressure pump. The mobile phase containing the analytes permeates through the stationary phase bed in a definite direction. The components of the mixture are separated depending on their chemical affinity with the mobile and stationary phases. The separation occurs after repeated sorption and desorption steps occurring when the liquid interacts with the stationary bed.[8] The liquid solvent (mobile phase) is delivered under high pressure (up to 400 bar or 5800 psi) into a packed column containing the stationary phase. The high pressure is necessary to achieve a constant flow rate for reproducible chromatography experiments. Depending on the partitioning between the mobile and stationary phases, the components of the sample will flow out of the column at different times.[16] The column is the most important component of the LC system and is designed to withstand the high pressure of the liquid. Conventional LC columns are 100–300 mm long with outer diameter of 6.4 mm (1/4 inch) and internal diameter of 3.0–4.6 mm. For applications involving LC-MS, the length of chromatography columns can be shorter (30–50 mm) with 3–5 μm diameter packing particles. In addition to the conventional model, other LC columns are the narrow bore, microbore, microcapillary, and nano-LC models. These columns have smaller internal diameters, allow for a more efficient separation, and handle liquid flows under 1 ml/min (the conventional flow-rate).[8] In order to improve separation efficiency and peak resolution, ultra performance liquid chromatography (UHPLC) can be used instead of HPLC. This LC variant uses columns packed with smaller silica particles (~1.7 μm diameter) and requires higher operating pressures in the range of 310000 to 775000 torr (6000 to 15000 psi, 400 to 1034 bar).[5]
Mass spectrometry[edit]

LC-MS spectrum of each resolved peak
Mass spectrometry (MS) is an analytical technique that measures the mass-to-charge ratio (m/z) of charged particles (ions). Although there are many different kinds of mass spectrometers, all of them make use of electric or magnetic fields to manipulate the motion of ions produced from an analyte of interest and determine their m/z.[18] The basic components of a mass spectrometer are the ion source, the mass analyzer, the detector, and the data and vacuum systems. The ion source is where the components of a sample introduced in a MS system are ionized by means of electron beams, photon beams (UV lights), laser beams or corona discharge. In the case of electrospray ionization, the ion source moves ions that exist in liquid solution into the gas phase. The ion source converts and fragments the neutral sample molecules into gas-phase ions that are sent to the mass analyzer. While the mass analyzer applies the electric and magnetic fields to sort the ions by their masses, the detector measures and amplifies the ion current to calculate the abundances of each mass-resolved ion. In order to generate a mass spectrum that a human eye can easily recognize, the data system records, processes, stores, and displays data in a computer.[5]
The mass spectrum can be used to determine the mass of the analytes, their elemental and isotopic composition, or to elucidate the chemical structure of the sample.[5] MS is an experiment that must take place in gas phase and under vacuum (1.33 * 10−2 to 1.33 * 10−6 pascal). Therefore, the development of devices facilitating the transition from samples at higher pressure and in condensed phase (solid or liquid) into a vacuum system has been essential to develop MS as a potent tool for identification and quantification of organic compounds like peptides.[19] MS is now in very common use in analytical laboratories that study physical, chemical, or biological properties of a great variety of compounds. Among the many different kinds of mass analyzers, the ones that find application in LC-MS systems are the quadrupole, time-of-flight (TOF), ion traps, and hybrid quadrupole-TOF (QTOF) analyzers.[7]
Interfaces[edit]
The interface between a liquid phase technique (HPLC) with a continuously flowing eluate, and a gas phase technique carried out in a vacuum was difficult for a long time. The advent of electrospray ionization changed this. Currently, the most common LC-MS interfaces are electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI), and atmospheric pressure photo-ionization (APPI). These are newer MS ion sources that facilitate the transition from a high pressure environment (HPLC) to high vacuum conditions needed at the MS analyzer.[20][7] Although these interfaces are described individually, they can also be commercially available as dual ESI/APCI, ESI/APPI, or APCI/APPI ion sources.[8] Various deposition and drying techniques were used in the past (e.g., moving belts) but the most common of these was the off-line MALDI deposition.[21][22] A new approach still under development called direct-EI LC-MS interface, couples a nano HPLC system and an electron ionization equipped mass spectrometer.[23][24]
Electrospray ionization (ESI)[edit]
ESI interface for LC-MS systems was developed by Fenn and collaborators in 1988.[25] This ion source/ interface can be used for the analysis of moderately polar and even very polar molecules (e.g., metabolites, xenobiotics, peptides, nucleotides, polysaccharides). The liquid eluate coming out of the LC column is directed into a metal capillary kept at 3 to 5 kV and is nebulized by a high-velocity coaxial flow of gas at the tip of the capillary, creating a fine spray of charged droplets in front of the entrance to the vacuum chamber. To avoid contamination of the vacuum system by buffers and salts, this capillary is usually perpendicularly located at the inlet of the MS system, in some cases with a counter-current of dry nitrogen in front of the entrance through which ions are directed by the electric field. In some sources, rapid droplet evaporation and thus maximum ion emission is achieved by mixing an additional stream of hot gas with the spray plume in front of the vacuum entrance. In other sources, the droplets are drawn through a heated capillary tube as they enter the vacuum, promoting droplet evaporation and ion emission. These methods of increasing droplet evaporation now allow the use of liquid flow rates of 1 — 2 mL/min to be used while still achieving efficient ionisation[26] and high sensitivity. Thus while the use of 1 — 3 mm microbore columns and lower flow rates of 50 — 200 μl/min was commonly considered necessary for optimum operation, this limitation is no longer as important, and the higher column capacity of larger bore columns can now be advantageously employed with ESI LC-MS systems. Positively and negatively charged ions can be created by switching polarities, and it is possible to acquire alternate positive and negative mode spectra rapidly within the same LC run . While most large molecules (greater than MW 1500-2000) produce multiply charged ions in the ESI source, the majority of smaller molecules produce singly charged ions.[7]
Atmospheric pressure chemical ionization (APCI)[edit]
The development of the APCI interface for LC-MS started with Horning and collaborators in the early 1973.[27] However, its commercial application was introduced at the beginning of the 1990s after Henion and collaborators improved the LC-APCI-MS interface in 1986.[8] The APCI ion source/ interface can be used to analyze small, neutral, relatively non-polar, and thermally stable molecules (e.g., steroids, lipids, and fat soluble vitamins). These compounds are not well ionized using ESI. In addition, APCI can also handle mobile phase streams containing buffering agents. The liquid from the LC system is pumped through a capillary and there is also nebulization at the tip, where a corona discharge takes place. First, the ionizing gas surrounding the interface and the mobile phase solvent are subject to chemical ionization at the ion source. Later, these ions react with the analyte and transfer their charge. The sample ions then pass through small orifice skimmers by means of or ion-focusing lenses. Once inside the high vacuum region, the ions are subject to mass analysis. This interface can be operated in positive and negative charge modes and singly-charged ions are mainly produced.[7] APCI ion source can also handle flow rates between 500 and 2000 μl/min and it can be directly connected to conventional 4.6 mm ID columns.[16]
Atmospheric pressure photoionization (APPI)[edit]
The APPI interface for LC-MS was developed simultaneously by Bruins and Syage in 2000.[28][8] APPI is another LC-MS ion source/ interface for the analysis of neutral compounds that cannot be ionized using ESI.[7] This interface is similar to the APCI ion source, but instead of a corona discharge, the ionization occurs by using photons coming from a discharge lamp. In the direct-APPI mode, singly charged analyte molecular ions are formed by absorption of a photon and ejection of an electron. In the dopant-APPI mode, an easily ionizable compound (Dopant) is added to the mobile phase or the nebulizing gas to promote a reaction of charge-exchange between the dopant molecular ion and the analyte. The ionized sample is later transferred to the mass analyzer at high vacuum as it passes through small orifice skimmers.[8]
Applications[edit]
The coupling of MS with LC systems is attractive because liquid chromatography can separate delicate and complex natural mixtures, which chemical composition needs to be well established (e.g., biological fluids, environmental samples, and drugs). Further, LC-MS has applications in volatile explosive residue analysis.[29] Nowadays, LC-MS has become one of the most widely used chemical analysis techniques because more than 85% of natural chemical compounds are polar and thermally labile and GC-MS cannot process these samples.[5] As an example, HPLC-MS is regarded as the leading analytical technique for proteomics and pharmaceutical laboratories.[7][5] Other important applications of LC-MS include the analysis of food, pesticides, and plant phenols.[8]
Pharmacokinetics[edit]
LC-MS is widely used in the field of bioanalysis and is specially involved in pharmacokinetic studies of pharmaceuticals. Pharmacokinetic studies are needed to determine how quickly a drug will be cleared from the body organs and the hepatic blood flow. MS analyzers are useful in these studies because of their shorter analysis time, and higher sensitivity and specificity compared to UV detectors commonly attached to HPLC systems. One major advantage is the use of tandem MS-MS, where the detector may be programmed to select certain ions to fragment. The measured quantity is the sum of molecule fragments chosen by the operator. As long as there are no interferences or ion suppression in LC-MS, the LC separation can be quite quick.[30]
Proteomics/metabolomics[edit]
LC-MS is used in proteomics as a method to detect and identify the components of a complex mixture. The bottom-up proteomics LC-MS approach generally involves protease digestion and denaturation using trypsin as a protease, urea to denature the tertiary structure, and iodoacetamide to modify the cysteine residues. After digestion, LC-MS is used for peptide mass fingerprinting, or LC-MS/MS (tandem MS) is used to derive the sequences of individual peptides.[31] LC-MS/MS is most commonly used for proteomic analysis of complex samples where peptide masses may overlap even with a high-resolution mass spectrometry. Samples of complex biological (e.g., human serum) may be analyzed in modern LC-MS/MS systems, which can identify over 1000 proteins. However, this high level of protein identification is possible only after separating the sample by means of SDS-PAGE gel or HPLC-SCX.[30] Recently, LC-MS/MS has been applied to search peptide biomarkers. Examples are the recent discovery and validation of peptide biomarkers for four major bacterial respiratory tract pathogens (Staphylococcus aureus, Moraxella catarrhalis; Haemophilus influenzae and Streptococcus pneumoniae) and the SARS-CoV-2 virus.[32] [33]
LC-MS has emerged as one of the most commonly used techniques in global metabolite profiling of biological tissue (e.g., blood plasma, serum, urine).[34] LC-MS is also used for the analysis of natural products and the profiling of secondary metabolites in plants.[35] In this regard, MS-based systems are useful to acquire more detailed information about the wide spectrum of compounds from a complex biological samples. LC-Nuclear magnetic resonance (NMR) is also used in plant metabolomics, but this technique can only detect and quantify the most abundant metabolites. LC-MS has been useful to advance the field of plant metabolomics, which aims to study the plant system at molecular level providing a non-biased characterization of the plant metabolome in response to its environment.[36] The first application of LC-MS in plant metabolomics was the detection of a wide range of highly polar metabolites, oligosaccharides, amino acids, amino sugars, and sugar nucleotides from Cucurbita maxima phloem tissues.[37] Another example of LC-MS in plant metabolomics is the efficient separation and identification of glucose, sucrose, raffinose, stachyose, and verbascose from leaf extracts of Arabidopsis thaliana.[38]
Drug development[edit]
LC-MS is frequently used in drug development because it allows quick molecular weight confirmation and structure identification. These features speed up the process of generating, testing, and validating a discovery starting from a vast array of products with potential application. LC-MS applications for drug development are highly automated methods used for peptide mapping, glycoprotein mapping, lipodomics, natural products dereplication, bioaffinity screening, in vivo drug screening, metabolic stability screening, metabolite identification, impurity identification, quantitative bioanalysis, and quality control.[39]
See also[edit]
- Gas chromatography–mass spectrometry
- Capillary electrophoresis–mass spectrometry
- Ion-mobility spectrometry–mass spectrometry
References[edit]
- ^ de Hoffmann, Edmond; Stroobant, Vincent (2002). Mass Spectrometry (Principles and Applications) (2nd ed.). Wiley. pp. 157–158. ISBN 0-471-48566-7.
- ^ Pitt, James J (February 2009). «Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry». Clin Biochem Rev. 30 (1): 19–34. PMC 2643089. PMID 19224008.
- ^ Zhou, Bin; Xiao, Jun Feng; Tuli, Leepika; Ressom, Habtom W (Feb 2012). «LC-MS-based metabolomics». Mol. Biosyst. 8 (2): 470–481. doi:10.1039/c1mb05350g. PMC 3699692. PMID 22041788.
- ^ Chaimbault, Patrick (2014-01-01). «The Modern Art of Identification of Natural Substances in Whole Plants». In Jacob, Claus; Kirsch, Gilbert; Slusarenko, Alan; Winyard, Paul G.; Burkholz, Torsten (eds.). Recent Advances in Redox Active Plant and Microbial Products. Springer Netherlands. pp. 31–94. doi:10.1007/978-94-017-8953-0_3. ISBN 9789401789523.
- ^ a b c d e f g h i j k l Dass, Chhabil (2007-01-01). «Hyphenated Separation Techniques». Fundamentals of Contemporary Mass Spectrometry. John Wiley & Sons, Inc. pp. 151–194. doi:10.1002/9780470118498.ch5. ISBN 9780470118498.
- ^ Seger, Christoph; Salzmann, Linda (2020-08-01). «After another decade: LC–MS/MS became routine in clinical diagnostics». Clinical Biochemistry. Advancement and Applications of Mass Spectrometry in Laboratory Medicine. 82: 2–11. doi:10.1016/j.clinbiochem.2020.03.004. ISSN 0009-9120. PMID 32188572. S2CID 213186669.
- ^ a b c d e f g h i j Pitt, James J (2017-03-12). «Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry». The Clinical Biochemist Reviews. 30 (1): 19–34. ISSN 0159-8090. PMC 2643089. PMID 19224008.
- ^ a b c d e f g h i j k l m n Niessen, Wilfried M. A (2006). Liquid Chromatography-Mass Spectrometry, Third Edition. Boca Raton: CRC Taylor & Francis. pp. 50–90. ISBN 9780824740825. OCLC 232370223.
- ^ James, A. T.; Martin, A. J. P. (1952-03-01). «Gas-liquid partition chromatography: the separation and micro-estimation of volatile fatty acids from formic acid to dodecanoic acid». Biochemical Journal. 50 (5): 679–690. doi:10.1042/bj0500679. ISSN 0264-6021. PMC 1197726. PMID 14934673.
- ^ Tal’roze, V. L; Karpov, G. V.; Gordetskii, I. G.; Skurat, V. E. (1968). «Capillary Systems for the Introduction of Liquid Mixtures into an Analytical Mass Spectrometer». Russ. J. Phys. Chem. 42: 1658–1664.
- ^ a b c d Arpino, Patrick (2006). «History of LC-MS Development and Interfacing». Encyclopedia of Mass Spectrometry. Vol. 8. Elsevier. ISBN 978-0080438474.
- ^ Tal’roze, V.L.; Gorodetskii, I.G.; Zolotoy, N.B; Karpov, G.V.; Skurat, V.E.; Maslennikova, V.Ya. (1978). «Capillary system for continuous introducing of volatile liquids into analytical MS and its application». Adv. Mass Spectrom. 7: 858.
- ^ Baldwin, M. A.; McLafferty, F. W. (1973). «Liquid chromatography-mass spectrometry interface-I: The direct introduction of liquid solutions into a chemical ionization mass spectrometer». Organic Mass Spectrometry. 7 (9): 1111–1112. doi:10.1002/oms.1210070913. ISSN 0030-493X.
- ^ Pullen, Franl (2010). «The fascinating history of the development of LC-MS; a personal perspective». Chromatography Today (February/March): 4–6.
- ^ de Koster, Chris G.; Schoenmakers, Peter J. (2020). «Chapter 3.1 — History of Liquid Chromatography-Mass Spectrometry». In Tranchida, Peter Q.; Mondello, Luigi (eds.). Hyphenations of Capillary Chromatography With Mass Spectrometry. Elsevier. pp. 279–295. ISBN 978-0-12-809638-3.
- ^ a b c d e Ardrey, Robert E. (2003-01-01). «Introduction». Liquid Chromatography – Mass Spectrometry: An Introduction. Analytical Techniques in the Sciences (AnTS). John Wiley & Sons, Ltd. pp. 1–5. doi:10.1002/0470867299.ch1. ISBN 9780470867297.
- ^ Blakley, C. R.; Carmody, J. J.; Vestal, M. L. (1980). «A New Soft Ionization Technique for Mass Spectrometry of Complex Samples». J. Am. Chem. Soc. 102: 5931–5933. doi:10.1021/ja00538a050.
- ^ Roberts, Gordon (2013). Roberts, Gordon C. K (ed.). Encyclopedia of Biophysics — Springer. doi:10.1007/978-3-642-16712-6. ISBN 978-3-642-16711-9. S2CID 44856071.
- ^ Sharp, Thomas R. (2009-01-01). «Mass Spectrometry». In Nassar, Ala F.; Collegiateessor, Paul F. Hollenberg; VP, JoAnn Scatina (eds.). Drug Metabolism Handbook. John Wiley & Sons, Inc. pp. 167–227. doi:10.1002/9780470439265.ch8. ISBN 9780470439265.
- ^ Arpino, Patrick (1992). «Combined liquid chromatography mass spectrometry. Part III. Applications of thermospray». Mass Spectrometry Reviews. 11 (1): 3–40. Bibcode:1992MSRv…11….3A. doi:10.1002/mas.1280110103.
- ^ Arpino, Patrick (1989). «Combined liquid chromatography mass spectrometry. Part I. Coupling by means of a moving belt interface». Mass Spectrometry Reviews. 8 (1): 35–55. Bibcode:1989MSRv….8…35A. doi:10.1002/mas.1280080103.
- ^ Murray, Kermit K. (1997). «Coupling matrix-assisted laser desorption/ionization to liquid separations». Mass Spectrometry Reviews. 16 (5): 283–299. Bibcode:1997MSRv…16..283M. doi:10.1002/(SICI)1098-2787(1997)16:5<283::AID-MAS3>3.0.CO;2-D.
- ^ Cappiello, Achille; Famiglini, Giorgio; Palma, Pierangela; Pierini, Elisabetta; Termopoli, Veronica; Trufelli, Helga (2008-12-01). «Overcoming Matrix Effects in Liquid Chromatography−Mass Spectrometry». Analytical Chemistry. 80 (23): 9343–9348. doi:10.1021/ac8018312. ISSN 0003-2700. PMID 19551950.
- ^ Cappiello, Achille; Famiglini, Giorgio; Mangani, Filippo; Palma, Pierangela (2002-03-01). «A simple approach for coupling liquid chromatography and electron ionization mass spectrometry». Journal of the American Society for Mass Spectrometry. 13 (3): 265–273. doi:10.1016/S1044-0305(01)00363-4. ISSN 1044-0305. PMID 11908806.
- ^ Fenn, J. B.; Mann, M.; Meng, C. K.; Wong, S. F.; Whitehouse, C. M. (1989-10-06). «Electrospray ionization for mass spectrometry of large biomolecules». Science. 246 (4926): 64–71. Bibcode:1989Sci…246…64F. CiteSeerX 10.1.1.522.9458. doi:10.1126/science.2675315. ISSN 0036-8075. PMID 2675315.
- ^ Covey, Tom (2022-08-30). «Where have all the ions gone, long time passing? Tandem quadrupole mass spectrometers with atmospheric pressure ionization sensitivity gains since the mid‐1970s. A perspective». Rapid Communications in Mass Spectrometry: e9354. doi:10.1002/rcm.9354. ISSN 0951-4198. PMID 35830299. S2CID 250491726.
- ^ Horning, E. C.; Horning, M. G.; Carroll, D. I.; Dzidic, I.; Stillwell, R. N. (1973-05-01). «New picogram detection system based on a mass spectrometer with an external ionization source at atmospheric pressure». Analytical Chemistry. 45 (6): 936–943. doi:10.1021/ac60328a035. ISSN 0003-2700.
- ^ Robb, null; Covey, null; Bruins, null (2000-08-01). «Atmospheric pressure photoionization: an ionization method for liquid chromatography-mass spectrometry». Analytical Chemistry. 72 (15): 3653–3659. doi:10.1021/ac0001636. ISSN 1520-6882. PMID 10952556.
- ^ Widmer, Leo; Watson, Stuart; Schlatter, Konrad; Crowson, Andrew (2002). «Development of an LC/MS method for the trace analysis of triacetone triperoxide (TATP)». The Analyst. 127 (12): 1627–1632. Bibcode:2002Ana…127.1627W. doi:10.1039/b208350g. ISSN 0003-2654. PMID 12537371.
- ^ a b Sudhakar, P.; Latha, P.; Reddy, P. V. (2016-04-05). Phenotyping Crop Plants for Physiological and Biochemical Traits. Academic Press. ISBN 9780128041109.
- ^ Wysocki VH, Resing KA, Zhang Q, Cheng G (2005). «Mass spectrometry of peptides and proteins». Methods. 35 (3): 211–22. doi:10.1016/j.ymeth.2004.08.013. PMID 15722218.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Karlsson, Roger; Thorsell, Annika; Gomila, Margarita; Salvà-Serra, Francisco; Jakobsson, Hedvig E.; Gonzales-Siles, Lucia; Jaén-Luchoro, Daniel; Skovbjerg, Susann; Fuchs, Johannes; Karlsson, Anders; Boulund, Fredrik (2020-03-01). «Discovery of Species-unique Peptide Biomarkers of Bacterial Pathogens by Tandem Mass Spectrometry-based Proteotyping». Molecular & Cellular Proteomics. 19 (3): 518–528. doi:10.1074/mcp.RA119.001667. ISSN 1535-9476. PMC 7050107. PMID 31941798.
- ^ Van Puyvelde, B; Van Uytfanghe, K; Tytgat, O; Van Oudenhove, L; Gabriels, R; Bouwmeester, R; Daled, S; Van Den Bossche, T; Ramasamy, P; Verhelst, S; De Clerck, L; Corveleyn, L; Willems, S; Debunne, N; Wynendaele, E; De Spiegeleer, B; Judak, P; Roels, K; De Wilde, L; Van Eenoo, P; Reyns, T; Cherlet, M; Dumont, E; Debyser, G; t’Kindt, R; Sandra, K; Gupta, S; Drouin, N; Harms, A; Hankemeier, T; Jones, DJL; Gupta, P; Lane, D; Lane, CS; El Ouadi, S; Vincendet, JB; Morrice, N; Oehrle, S; Tanna, N; Silvester, S; Hannam, S; Sigloch, FC; Bhangu-Uhlmann, A; Claereboudt, J; Anderson, NL; Razavi, M; Degroeve, S; Cuypers, L; Stove, C; Lagrou, K; Martens, GA; Deforce, D; Martens, L; Vissers, JPC; Dhaenens, M (28 June 2021). «Cov-MS: A Community-Based Template Assay for Mass-Spectrometry-Based Protein Detection in SARS-CoV-2 Patients». JACS Au. 1 (6): 750–765. doi:10.1021/jacsau.1c00048. PMC 8230961. PMID 34254058.
- ^ Gika, Helen G.; Theodoridis, Georgios A.; Plumb, Robert S.; Wilson, Ian D. (January 2014). «Current practice of liquid chromatography–mass spectrometry in metabolomics and metabonomics». Journal of Pharmaceutical and Biomedical Analysis. 87: 12–25. doi:10.1016/j.jpba.2013.06.032. ISSN 0731-7085. PMID 23916607.
- ^ Stobiecki, M.; Skirycz, A.; Kerhoas, L.; Kachlicki, P.; Muth, D.; Einhorn, J.; Mueller-Roeber, B. (2006). «Profiling of phenolic glycosidic conjugates in leaves of Arabidopsis thaliana using LC/MS». Metabolomics. 2 (4): 197–219. doi:10.1007/s11306-006-0031-5. S2CID 39140266.
- ^ Jorge, Tiago F.; Rodrigues, João A.; Caldana, Camila; Schmidt, Romy; van Dongen, Joost T.; Thomas-Oates, Jane; António, Carla (2016-09-01). «Mass spectrometry-based plant metabolomics: Metabolite responses to abiotic stress». Mass Spectrometry Reviews. 35 (5): 620–649. Bibcode:2016MSRv…35..620J. doi:10.1002/mas.21449. ISSN 1098-2787. PMID 25589422.
- ^ Tolstikov, Vladimir V.; Fiehn, Oliver (2002). «Analysis of Highly Polar Compounds of Plant Origin: Combination of Hydrophilic Interaction Chromatography and Electrospray Ion Trap Mass Spectrometry». Analytical Biochemistry. 301 (2): 298–307. doi:10.1006/abio.2001.5513. PMID 11814300. S2CID 3156968.
- ^ Antonio, Carla; Larson, Tony; Gilday, Alison; Graham, Ian; Bergström, Ed; Thomas-Oates, Jane (2008). «Hydrophilic interaction chromatography/electrospray mass spectrometry analysis of carbohydrate-related metabolites from Arabidopsis thaliana leaf tissue». Rapid Communications in Mass Spectrometry. 22 (9): 1399–1407. Bibcode:2008RCMS…22.1399A. doi:10.1002/rcm.3519. PMID 18384194.
- ^ Lee, Mike S.; Kerns, Edward H. (1999). «LC/MS applications in drug development». Mass Spectrometry Reviews. 18 (3–4): 187–279. Bibcode:1999MSRv…18..187L. doi:10.1002/(SICI)1098-2787(1999)18:3/4<187::AID-MAS2>3.0.CO;2-K. PMID 10568041.
Further reading[edit]
- Thurman, E. M.; Ferrer, Imma (2003). Liquid chromatography/mass spectrometry, MS/MS and time of flight MS: analysis of emerging contaminants. Columbus, OH: American Chemical Society. ISBN 978-0-8412-3825-1.
- Ferrer, Imma; Thurman, E. M. (2009). Liquid chromatography-Time of Flight Mass Spectrometry: Principles, Tools and Applications for Accurate Mass Analysis. New York, NJ: Wiley. ISBN 978-0-470-13797-0.
- McMaster, Marvin C. (2005). LC/MS: a practical user’s guide. New York: John Wiley. ISBN 978-0-471-65531-2.
- Yergey, Alfred L. (1990). Liquid chromatography/mass spectrometry: techniques and applications. New York: Plenum Press. ISBN 978-0-306-43186-9.
|
Ion trap LCMS system with ESI interface |
|
| Acronym | LCMS |
|---|---|
| Classification | Chromatography Mass spectrometry |
| Analytes | organic molecules biomolecules |
| Manufacturers | Agilent Bruker PerkinElmer SCIEX Shimadzu Scientific Thermo Fisher Scientific Waters Corporation |
| Other techniques | |
| Related | Gas chromatography–mass spectrometry |
Liquid chromatography–mass spectrometry (LC–MS) is an analytical chemistry technique that combines the physical separation capabilities of liquid chromatography (or HPLC) with the mass analysis capabilities of mass spectrometry (MS). Coupled chromatography — MS systems are popular in chemical analysis because the individual capabilities of each technique are enhanced synergistically. While liquid chromatography separates mixtures with multiple components, mass spectrometry provides spectral information that may help to identify (or confirm the suspected identity of) each separated component.[1] MS is not only sensitive, but provides selective detection, relieving the need for complete chromatographic separation.[2] LC-MS is also appropriate for metabolomics because of its good coverage of a wide range of chemicals.[3] This tandem technique can be used to analyze biochemical, organic, and inorganic compounds commonly found in complex samples of environmental and biological origin. Therefore, LC-MS may be applied in a wide range of sectors including biotechnology, environment monitoring, food processing, and pharmaceutical, agrochemical, and cosmetic industries.[4][5] Since the early 2000s, LC-MS (or more specifically LC-MS-MS) has also begun to be used in clinical applications.[6]
In addition to the liquid chromatography and mass spectrometry devices, an LC-MS system contains an interface that efficiently transfers the separated components from the LC column into the MS ion source.[5][7] The interface is necessary because the LC and MS devices are fundamentally incompatible. While the mobile phase in a LC system is a pressurized liquid, the MS analyzers commonly operate under high vacuum. Thus, it is not possible to directly pump the eluate from the LC column into the MS source. Overall, the interface is a mechanically simple part of the LC-MS system that transfers the maximum amount of analyte, removes a significant portion of the mobile phase used in LC and preserves the chemical identity of the chromatography products (chemically inert). As a requirement, the interface should not interfere with the ionizing efficiency and vacuum conditions of the MS system.[5] Nowadays, most extensively applied LC-MS interfaces are based on atmospheric pressure ionization (API) strategies like electrospray ionization (ESI), atmospheric-pressure chemical ionization (APCI), and atmospheric pressure photoionization (APPI). These interfaces became available in the 1990s after a two decade long research and development process.[8][7]
History of LC-MS[edit]
The coupling of chromatography with MS is a well developed chemical analysis strategy dating back from the 1950s. Gas chromatography (GC)–MS was originally introduced in 1952, when A. T. James and A. J. P. Martin were trying to develop tandem separation — mass analysis techniques.[9] In GC, the analytes are eluted from the separation column as a gas and the connection with electron ionization (EI) or chemical ionization (CI) ion sources in the MS system was a technically simpler challenge. Because of this, the development of GC-MS systems was faster than LC-MS and such systems were first commercialized in the 1970s.[7] The development of LC-MS systems took longer than GC-MS and was directly related to the development of proper interfaces. V. L. Tal’roze and collaborators started the development of LC-MS in the late 1960s,[10][11] when they first used capillaries to connect an LC columns to an EI source.[12][8] A similar strategy was investigated by McLafferty and collaborators in 1973 who coupled the LC column to a CI source,[13] which allowed a higher liquid flow into the source. This was the first and most obvious way of coupling LC with MS, and was known as the capillary inlet interface. This pioneer interface for LC-MS had the same analysis capabilities of GC-MS and was limited to rather volatile analytes and non-polar compounds with low molecular mass (below 400 Da). In the capillary inlet interface, the evaporation of the mobile phase inside the capillary was one of the main issues. Within the first years of development of LC-MS, on-line and off-line alternatives were proposed as coupling alternatives. In general, off-line coupling involved fraction collection, evaporation of solvent, and transfer of analytes to the MS using probes. Off-line analyte treatment process was time consuming and there was an inherent risk of sample contamination. Rapidly, it was realized that the analysis of complex mixtures would require the development of a fully automated on-line coupling solution in LC-MS.[8]
The key to the success and wide-spread adoption of LC-MS as a routine analytical tool lies in the interface and ion source between the liquid-based LC and the vacuum-base MS. The following interfaces were stepping-stones on the way to the modern atmospheric-pressure ionization interfaces, and are described for historical interest.
Moving Belt Interface[edit]
The moving-belt interface (MBI) was developed by McFadden et al in 1977 and commercialized by Finnigan.[14] This interface consisted of an endless moving belt onto which the LC column effluent was deposited in a band. On the belt, the solvent was evaporated by gently heating and efficiently exhausting the solvent vapours under reduced pressure in two vacuum chambers. After the liquid phase was removed, the belt passed over a heater which flash desorbed the analytes into the MS ion source. One of the significant advantages of the MBI was its compatibility with a wide range of chromatographic conditions.[8] MBI was successfully used for LC-MS applications between 1978 and 1990 because it allowed coupling of LC to MS devices using EI, CI, and fast-atom bombardment (FAB) ion sources. The most common MS systems connected by MBI interfaces to LC columns were magnetic sector and quadrupole instruments. MBI interfaces for LC-MS allowed MS to be widely applied in the analysis of drugs, pesticides, steroids, alkaloids, and polycyclic aromatic hydrocarbons. This interface is no longer used because of its mechanical complexity and the difficulties associated with belt renewal as well as its inability to handle very labile biomolecules.
Direct liquid introduction interface[edit]
The direct liquid introduction (DLI) interface was developed in 1980. This interface was intended to solve the problem of evaporation of liquid inside the capillary inlet interface. In DLI, a small portion of the LC flow was forced through a small aperture or diaphragm (typically 10um in diameter) to form a liquid jet composed of small droplets that were subsequently dried in a desolvation chamber.[11] The analytes were ionized using a solvent assisted chemical ionization source, where the LC solvents acted as reagent gases. To use this interface, it was necessary to split the flow coming out of the LC column because only a small portion of the effluent (10 to 50 μl/min out of 1 ml/min) could be introduced into the source without raising the vacuum pressure of the MS system too high. Alternately, Henion at Cornell University had success with using micro-bore LC methods so that the entire (low) flow of the LC could be used. One of the main operational problems of the DLI interface was the frequent clogging of the diaphragm orifices. The DLI interface was used between 1982 and 1985 for the analysis of pesticides, corticosteroids, metabolites in horse urine, erythromycin, and vitamin B12. However, this interface was replaced by the thermospray interface, which removed the flow rate limitations and the issues with the clogging diaphragms.[5][8]
A related device was the particle beam interface (PBI), developed by Willoughby and Browner in 1984.[15] Particle beam interfaces took over the wide applications of MBI for LC-MS in 1988.[8][16] The PBI operated by using a helium gas nebulizer to spray the eluant into the vacuum, drying the droplets and pumping away the solvent vapour (using a jet separator) while the stream of monodisperse dried particles containing the analyte entered the source.[11] Drying the droplets outside of the source volume, and using a jet separator to pump away the solvent vapour, allowed the particles to enter and be vapourized in a low-pressure EI source. As with the MBI, the ability to generate library-searchable EI spectra was a distinct advantage for many applications. Commercialized by Hewlett Packard, and later by VG and Extrel, it enjoyed moderate success, but has been largely supplanted by the atmospheric pressure interfaces such as electrospray and APCI which provide a broader range of compound coverage and applications.
Thermospray interface[edit]
The thermospray (TSP) interface was developed in 1980 by Marvin Vestal and co-workers at the University of Houston.[17] It was commercialized by Vestec and several of the major mass spectrometer manufacurers. The interface resulted from a long term research project intended to find a LC-MS interface capable of handling high flow rates (1 ml/min) and avoiding the flow split in DLI interfaces. The TSP interface was composed of a heated probe, a desolvation chamber, and an ion focusing skimmer. The LC effluent passed through the heated probe and emerged as a jet of vapor and small droplets flowing into the desolvation chamber at low pressure. Initially operated with a filament or discharge as the source of ions (thereby acting as a CI source for vapourized analyte), it was soon discovered that ions were also observed when the filament or discharge was off. This could be attributed to either direct emission of ions from the liquid droplets as they evaporated in a process related to electrospray ionization or ion evaporation, or to chemical ionization of vapourized analyte molecules from buffer ions (such as ammonium acetate). The fact that multiply-charged ions were observed from some larger analytes suggests that direct analyte ion emission was occurring under at least some conditions.[11] The interface was able to handle up to 2 ml/min of eluate from the LC column and would efficiently introduce it into the MS vacuum system. TSP was also more suitable for LC-MS applications involving reversed phase liquid chromatography (RT-LC). With time, the mechanical complexity of TSP was simplified, and this interface became popular as the first ideal LC-MS interface for pharmaceutical applications comprising the analysis of drugs, metabolites, conjugates, nucleosides, peptides, natural products, and pesticides. The introduction of TSP marked a significant improvement for LC-MS systems and was the most widely applied interface until the beginning of the 1990s, when it began to be replaced by interfaces involving atmospheric pressure ionization (API).[5][7][16]
FAB based interfaces[edit]
The frit fast atom bombardment (FAB) and continuous flow-FAB (CF-FAB) interfaces were developed in 1985 and 1986 respectively.[16] Both interfaces were similar, but they differed in that the first used a porous frit probe as connecting channel, while CF-FAB used a probe tip. From these, the CF-FAB was more successful as a LC-MS interface and was useful to analyze non-volatile and thermally labile compounds. In these interfaces, the LC effluent passed through the frit or CF-FAB channels to form a uniform liquid film at the tip. There, the liquid was bombarded with ion beams or high energy atoms (fast atoms). For stable operation, the FAB based interfaces were able to handle liquid flow rates of only 1–15 μl and were also restricted to microbore and capillary columns. In order to be used in FAB MS ionization sources, the analytes of interest had to be mixed with a matrix (e.g., glycerol) that could be added before or after the separation in the LC column. FAB based interfaces were extensively used to characterize peptides, but lost applicability with the advent of electrospray based interfaces in 1988.[5][8]
Liquid chromatography[edit]
Diagram of an LC-MS system
Liquid chromatography is a method of physical separation in which the components of a liquid mixture are distributed between two immiscible phases, i.e., stationary and mobile. The practice of LC can be divided into five categories, i.e., adsorption chromatography, partition chromatography, ion-exchange chromatography, size-exclusion chromatography, and affinity chromatography. Among these, the most widely used variant is the reverse-phase (RP) mode of the partition chromatography technique, which makes use of a nonpolar (hydrophobic) stationary phase and a polar mobile phase. In common applications, the mobile phase is a mixture of water and other polar solvents (e.g., methanol, isopropanol, and acetonitrile), and the stationary matrix is prepared by attaching long-chain alkyl groups (e.g., n-octadecyl or C18) to the external and internal surfaces of irregularly or spherically shaped 5 μm diameter porous silica particles.[5]
In HPLC, typically 20 μl of the sample of interest are injected into the mobile phase stream delivered by a high pressure pump. The mobile phase containing the analytes permeates through the stationary phase bed in a definite direction. The components of the mixture are separated depending on their chemical affinity with the mobile and stationary phases. The separation occurs after repeated sorption and desorption steps occurring when the liquid interacts with the stationary bed.[8] The liquid solvent (mobile phase) is delivered under high pressure (up to 400 bar or 5800 psi) into a packed column containing the stationary phase. The high pressure is necessary to achieve a constant flow rate for reproducible chromatography experiments. Depending on the partitioning between the mobile and stationary phases, the components of the sample will flow out of the column at different times.[16] The column is the most important component of the LC system and is designed to withstand the high pressure of the liquid. Conventional LC columns are 100–300 mm long with outer diameter of 6.4 mm (1/4 inch) and internal diameter of 3.0–4.6 mm. For applications involving LC-MS, the length of chromatography columns can be shorter (30–50 mm) with 3–5 μm diameter packing particles. In addition to the conventional model, other LC columns are the narrow bore, microbore, microcapillary, and nano-LC models. These columns have smaller internal diameters, allow for a more efficient separation, and handle liquid flows under 1 ml/min (the conventional flow-rate).[8] In order to improve separation efficiency and peak resolution, ultra performance liquid chromatography (UHPLC) can be used instead of HPLC. This LC variant uses columns packed with smaller silica particles (~1.7 μm diameter) and requires higher operating pressures in the range of 310000 to 775000 torr (6000 to 15000 psi, 400 to 1034 bar).[5]
Mass spectrometry[edit]
LC-MS spectrum of each resolved peak
Mass spectrometry (MS) is an analytical technique that measures the mass-to-charge ratio (m/z) of charged particles (ions). Although there are many different kinds of mass spectrometers, all of them make use of electric or magnetic fields to manipulate the motion of ions produced from an analyte of interest and determine their m/z.[18] The basic components of a mass spectrometer are the ion source, the mass analyzer, the detector, and the data and vacuum systems. The ion source is where the components of a sample introduced in a MS system are ionized by means of electron beams, photon beams (UV lights), laser beams or corona discharge. In the case of electrospray ionization, the ion source moves ions that exist in liquid solution into the gas phase. The ion source converts and fragments the neutral sample molecules into gas-phase ions that are sent to the mass analyzer. While the mass analyzer applies the electric and magnetic fields to sort the ions by their masses, the detector measures and amplifies the ion current to calculate the abundances of each mass-resolved ion. In order to generate a mass spectrum that a human eye can easily recognize, the data system records, processes, stores, and displays data in a computer.[5]
The mass spectrum can be used to determine the mass of the analytes, their elemental and isotopic composition, or to elucidate the chemical structure of the sample.[5] MS is an experiment that must take place in gas phase and under vacuum (1.33 * 10−2 to 1.33 * 10−6 pascal). Therefore, the development of devices facilitating the transition from samples at higher pressure and in condensed phase (solid or liquid) into a vacuum system has been essential to develop MS as a potent tool for identification and quantification of organic compounds like peptides.[19] MS is now in very common use in analytical laboratories that study physical, chemical, or biological properties of a great variety of compounds. Among the many different kinds of mass analyzers, the ones that find application in LC-MS systems are the quadrupole, time-of-flight (TOF), ion traps, and hybrid quadrupole-TOF (QTOF) analyzers.[7]
Interfaces[edit]
The interface between a liquid phase technique (HPLC) with a continuously flowing eluate, and a gas phase technique carried out in a vacuum was difficult for a long time. The advent of electrospray ionization changed this. Currently, the most common LC-MS interfaces are electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI), and atmospheric pressure photo-ionization (APPI). These are newer MS ion sources that facilitate the transition from a high pressure environment (HPLC) to high vacuum conditions needed at the MS analyzer.[20][7] Although these interfaces are described individually, they can also be commercially available as dual ESI/APCI, ESI/APPI, or APCI/APPI ion sources.[8] Various deposition and drying techniques were used in the past (e.g., moving belts) but the most common of these was the off-line MALDI deposition.[21][22] A new approach still under development called direct-EI LC-MS interface, couples a nano HPLC system and an electron ionization equipped mass spectrometer.[23][24]
Electrospray ionization (ESI)[edit]
ESI interface for LC-MS systems was developed by Fenn and collaborators in 1988.[25] This ion source/ interface can be used for the analysis of moderately polar and even very polar molecules (e.g., metabolites, xenobiotics, peptides, nucleotides, polysaccharides). The liquid eluate coming out of the LC column is directed into a metal capillary kept at 3 to 5 kV and is nebulized by a high-velocity coaxial flow of gas at the tip of the capillary, creating a fine spray of charged droplets in front of the entrance to the vacuum chamber. To avoid contamination of the vacuum system by buffers and salts, this capillary is usually perpendicularly located at the inlet of the MS system, in some cases with a counter-current of dry nitrogen in front of the entrance through which ions are directed by the electric field. In some sources, rapid droplet evaporation and thus maximum ion emission is achieved by mixing an additional stream of hot gas with the spray plume in front of the vacuum entrance. In other sources, the droplets are drawn through a heated capillary tube as they enter the vacuum, promoting droplet evaporation and ion emission. These methods of increasing droplet evaporation now allow the use of liquid flow rates of 1 — 2 mL/min to be used while still achieving efficient ionisation[26] and high sensitivity. Thus while the use of 1 — 3 mm microbore columns and lower flow rates of 50 — 200 μl/min was commonly considered necessary for optimum operation, this limitation is no longer as important, and the higher column capacity of larger bore columns can now be advantageously employed with ESI LC-MS systems. Positively and negatively charged ions can be created by switching polarities, and it is possible to acquire alternate positive and negative mode spectra rapidly within the same LC run . While most large molecules (greater than MW 1500-2000) produce multiply charged ions in the ESI source, the majority of smaller molecules produce singly charged ions.[7]
Atmospheric pressure chemical ionization (APCI)[edit]
The development of the APCI interface for LC-MS started with Horning and collaborators in the early 1973.[27] However, its commercial application was introduced at the beginning of the 1990s after Henion and collaborators improved the LC-APCI-MS interface in 1986.[8] The APCI ion source/ interface can be used to analyze small, neutral, relatively non-polar, and thermally stable molecules (e.g., steroids, lipids, and fat soluble vitamins). These compounds are not well ionized using ESI. In addition, APCI can also handle mobile phase streams containing buffering agents. The liquid from the LC system is pumped through a capillary and there is also nebulization at the tip, where a corona discharge takes place. First, the ionizing gas surrounding the interface and the mobile phase solvent are subject to chemical ionization at the ion source. Later, these ions react with the analyte and transfer their charge. The sample ions then pass through small orifice skimmers by means of or ion-focusing lenses. Once inside the high vacuum region, the ions are subject to mass analysis. This interface can be operated in positive and negative charge modes and singly-charged ions are mainly produced.[7] APCI ion source can also handle flow rates between 500 and 2000 μl/min and it can be directly connected to conventional 4.6 mm ID columns.[16]
Atmospheric pressure photoionization (APPI)[edit]
The APPI interface for LC-MS was developed simultaneously by Bruins and Syage in 2000.[28][8] APPI is another LC-MS ion source/ interface for the analysis of neutral compounds that cannot be ionized using ESI.[7] This interface is similar to the APCI ion source, but instead of a corona discharge, the ionization occurs by using photons coming from a discharge lamp. In the direct-APPI mode, singly charged analyte molecular ions are formed by absorption of a photon and ejection of an electron. In the dopant-APPI mode, an easily ionizable compound (Dopant) is added to the mobile phase or the nebulizing gas to promote a reaction of charge-exchange between the dopant molecular ion and the analyte. The ionized sample is later transferred to the mass analyzer at high vacuum as it passes through small orifice skimmers.[8]
Applications[edit]
The coupling of MS with LC systems is attractive because liquid chromatography can separate delicate and complex natural mixtures, which chemical composition needs to be well established (e.g., biological fluids, environmental samples, and drugs). Further, LC-MS has applications in volatile explosive residue analysis.[29] Nowadays, LC-MS has become one of the most widely used chemical analysis techniques because more than 85% of natural chemical compounds are polar and thermally labile and GC-MS cannot process these samples.[5] As an example, HPLC-MS is regarded as the leading analytical technique for proteomics and pharmaceutical laboratories.[7][5] Other important applications of LC-MS include the analysis of food, pesticides, and plant phenols.[8]
Pharmacokinetics[edit]
LC-MS is widely used in the field of bioanalysis and is specially involved in pharmacokinetic studies of pharmaceuticals. Pharmacokinetic studies are needed to determine how quickly a drug will be cleared from the body organs and the hepatic blood flow. MS analyzers are useful in these studies because of their shorter analysis time, and higher sensitivity and specificity compared to UV detectors commonly attached to HPLC systems. One major advantage is the use of tandem MS-MS, where the detector may be programmed to select certain ions to fragment. The measured quantity is the sum of molecule fragments chosen by the operator. As long as there are no interferences or ion suppression in LC-MS, the LC separation can be quite quick.[30]
Proteomics/metabolomics[edit]
LC-MS is used in proteomics as a method to detect and identify the components of a complex mixture. The bottom-up proteomics LC-MS approach generally involves protease digestion and denaturation using trypsin as a protease, urea to denature the tertiary structure, and iodoacetamide to modify the cysteine residues. After digestion, LC-MS is used for peptide mass fingerprinting, or LC-MS/MS (tandem MS) is used to derive the sequences of individual peptides.[31] LC-MS/MS is most commonly used for proteomic analysis of complex samples where peptide masses may overlap even with a high-resolution mass spectrometry. Samples of complex biological (e.g., human serum) may be analyzed in modern LC-MS/MS systems, which can identify over 1000 proteins. However, this high level of protein identification is possible only after separating the sample by means of SDS-PAGE gel or HPLC-SCX.[30] Recently, LC-MS/MS has been applied to search peptide biomarkers. Examples are the recent discovery and validation of peptide biomarkers for four major bacterial respiratory tract pathogens (Staphylococcus aureus, Moraxella catarrhalis; Haemophilus influenzae and Streptococcus pneumoniae) and the SARS-CoV-2 virus.[32] [33]
LC-MS has emerged as one of the most commonly used techniques in global metabolite profiling of biological tissue (e.g., blood plasma, serum, urine).[34] LC-MS is also used for the analysis of natural products and the profiling of secondary metabolites in plants.[35] In this regard, MS-based systems are useful to acquire more detailed information about the wide spectrum of compounds from a complex biological samples. LC-Nuclear magnetic resonance (NMR) is also used in plant metabolomics, but this technique can only detect and quantify the most abundant metabolites. LC-MS has been useful to advance the field of plant metabolomics, which aims to study the plant system at molecular level providing a non-biased characterization of the plant metabolome in response to its environment.[36] The first application of LC-MS in plant metabolomics was the detection of a wide range of highly polar metabolites, oligosaccharides, amino acids, amino sugars, and sugar nucleotides from Cucurbita maxima phloem tissues.[37] Another example of LC-MS in plant metabolomics is the efficient separation and identification of glucose, sucrose, raffinose, stachyose, and verbascose from leaf extracts of Arabidopsis thaliana.[38]
Drug development[edit]
LC-MS is frequently used in drug development because it allows quick molecular weight confirmation and structure identification. These features speed up the process of generating, testing, and validating a discovery starting from a vast array of products with potential application. LC-MS applications for drug development are highly automated methods used for peptide mapping, glycoprotein mapping, lipodomics, natural products dereplication, bioaffinity screening, in vivo drug screening, metabolic stability screening, metabolite identification, impurity identification, quantitative bioanalysis, and quality control.[39]
See also[edit]
- Gas chromatography–mass spectrometry
- Capillary electrophoresis–mass spectrometry
- Ion-mobility spectrometry–mass spectrometry
References[edit]
- ^ de Hoffmann, Edmond; Stroobant, Vincent (2002). Mass Spectrometry (Principles and Applications) (2nd ed.). Wiley. pp. 157–158. ISBN 0-471-48566-7.
- ^ Pitt, James J (February 2009). «Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry». Clin Biochem Rev. 30 (1): 19–34. PMC 2643089. PMID 19224008.
- ^ Zhou, Bin; Xiao, Jun Feng; Tuli, Leepika; Ressom, Habtom W (Feb 2012). «LC-MS-based metabolomics». Mol. Biosyst. 8 (2): 470–481. doi:10.1039/c1mb05350g. PMC 3699692. PMID 22041788.
- ^ Chaimbault, Patrick (2014-01-01). «The Modern Art of Identification of Natural Substances in Whole Plants». In Jacob, Claus; Kirsch, Gilbert; Slusarenko, Alan; Winyard, Paul G.; Burkholz, Torsten (eds.). Recent Advances in Redox Active Plant and Microbial Products. Springer Netherlands. pp. 31–94. doi:10.1007/978-94-017-8953-0_3. ISBN 9789401789523.
- ^ a b c d e f g h i j k l Dass, Chhabil (2007-01-01). «Hyphenated Separation Techniques». Fundamentals of Contemporary Mass Spectrometry. John Wiley & Sons, Inc. pp. 151–194. doi:10.1002/9780470118498.ch5. ISBN 9780470118498.
- ^ Seger, Christoph; Salzmann, Linda (2020-08-01). «After another decade: LC–MS/MS became routine in clinical diagnostics». Clinical Biochemistry. Advancement and Applications of Mass Spectrometry in Laboratory Medicine. 82: 2–11. doi:10.1016/j.clinbiochem.2020.03.004. ISSN 0009-9120. PMID 32188572. S2CID 213186669.
- ^ a b c d e f g h i j Pitt, James J (2017-03-12). «Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry». The Clinical Biochemist Reviews. 30 (1): 19–34. ISSN 0159-8090. PMC 2643089. PMID 19224008.
- ^ a b c d e f g h i j k l m n Niessen, Wilfried M. A (2006). Liquid Chromatography-Mass Spectrometry, Third Edition. Boca Raton: CRC Taylor & Francis. pp. 50–90. ISBN 9780824740825. OCLC 232370223.
- ^ James, A. T.; Martin, A. J. P. (1952-03-01). «Gas-liquid partition chromatography: the separation and micro-estimation of volatile fatty acids from formic acid to dodecanoic acid». Biochemical Journal. 50 (5): 679–690. doi:10.1042/bj0500679. ISSN 0264-6021. PMC 1197726. PMID 14934673.
- ^ Tal’roze, V. L; Karpov, G. V.; Gordetskii, I. G.; Skurat, V. E. (1968). «Capillary Systems for the Introduction of Liquid Mixtures into an Analytical Mass Spectrometer». Russ. J. Phys. Chem. 42: 1658–1664.
- ^ a b c d Arpino, Patrick (2006). «History of LC-MS Development and Interfacing». Encyclopedia of Mass Spectrometry. Vol. 8. Elsevier. ISBN 978-0080438474.
- ^ Tal’roze, V.L.; Gorodetskii, I.G.; Zolotoy, N.B; Karpov, G.V.; Skurat, V.E.; Maslennikova, V.Ya. (1978). «Capillary system for continuous introducing of volatile liquids into analytical MS and its application». Adv. Mass Spectrom. 7: 858.
- ^ Baldwin, M. A.; McLafferty, F. W. (1973). «Liquid chromatography-mass spectrometry interface-I: The direct introduction of liquid solutions into a chemical ionization mass spectrometer». Organic Mass Spectrometry. 7 (9): 1111–1112. doi:10.1002/oms.1210070913. ISSN 0030-493X.
- ^ Pullen, Franl (2010). «The fascinating history of the development of LC-MS; a personal perspective». Chromatography Today (February/March): 4–6.
- ^ de Koster, Chris G.; Schoenmakers, Peter J. (2020). «Chapter 3.1 — History of Liquid Chromatography-Mass Spectrometry». In Tranchida, Peter Q.; Mondello, Luigi (eds.). Hyphenations of Capillary Chromatography With Mass Spectrometry. Elsevier. pp. 279–295. ISBN 978-0-12-809638-3.
- ^ a b c d e Ardrey, Robert E. (2003-01-01). «Introduction». Liquid Chromatography – Mass Spectrometry: An Introduction. Analytical Techniques in the Sciences (AnTS). John Wiley & Sons, Ltd. pp. 1–5. doi:10.1002/0470867299.ch1. ISBN 9780470867297.
- ^ Blakley, C. R.; Carmody, J. J.; Vestal, M. L. (1980). «A New Soft Ionization Technique for Mass Spectrometry of Complex Samples». J. Am. Chem. Soc. 102: 5931–5933. doi:10.1021/ja00538a050.
- ^ Roberts, Gordon (2013). Roberts, Gordon C. K (ed.). Encyclopedia of Biophysics — Springer. doi:10.1007/978-3-642-16712-6. ISBN 978-3-642-16711-9. S2CID 44856071.
- ^ Sharp, Thomas R. (2009-01-01). «Mass Spectrometry». In Nassar, Ala F.; Collegiateessor, Paul F. Hollenberg; VP, JoAnn Scatina (eds.). Drug Metabolism Handbook. John Wiley & Sons, Inc. pp. 167–227. doi:10.1002/9780470439265.ch8. ISBN 9780470439265.
- ^ Arpino, Patrick (1992). «Combined liquid chromatography mass spectrometry. Part III. Applications of thermospray». Mass Spectrometry Reviews. 11 (1): 3–40. Bibcode:1992MSRv…11….3A. doi:10.1002/mas.1280110103.
- ^ Arpino, Patrick (1989). «Combined liquid chromatography mass spectrometry. Part I. Coupling by means of a moving belt interface». Mass Spectrometry Reviews. 8 (1): 35–55. Bibcode:1989MSRv….8…35A. doi:10.1002/mas.1280080103.
- ^ Murray, Kermit K. (1997). «Coupling matrix-assisted laser desorption/ionization to liquid separations». Mass Spectrometry Reviews. 16 (5): 283–299. Bibcode:1997MSRv…16..283M. doi:10.1002/(SICI)1098-2787(1997)16:5<283::AID-MAS3>3.0.CO;2-D.
- ^ Cappiello, Achille; Famiglini, Giorgio; Palma, Pierangela; Pierini, Elisabetta; Termopoli, Veronica; Trufelli, Helga (2008-12-01). «Overcoming Matrix Effects in Liquid Chromatography−Mass Spectrometry». Analytical Chemistry. 80 (23): 9343–9348. doi:10.1021/ac8018312. ISSN 0003-2700. PMID 19551950.
- ^ Cappiello, Achille; Famiglini, Giorgio; Mangani, Filippo; Palma, Pierangela (2002-03-01). «A simple approach for coupling liquid chromatography and electron ionization mass spectrometry». Journal of the American Society for Mass Spectrometry. 13 (3): 265–273. doi:10.1016/S1044-0305(01)00363-4. ISSN 1044-0305. PMID 11908806.
- ^ Fenn, J. B.; Mann, M.; Meng, C. K.; Wong, S. F.; Whitehouse, C. M. (1989-10-06). «Electrospray ionization for mass spectrometry of large biomolecules». Science. 246 (4926): 64–71. Bibcode:1989Sci…246…64F. CiteSeerX 10.1.1.522.9458. doi:10.1126/science.2675315. ISSN 0036-8075. PMID 2675315.
- ^ Covey, Tom (2022-08-30). «Where have all the ions gone, long time passing? Tandem quadrupole mass spectrometers with atmospheric pressure ionization sensitivity gains since the mid‐1970s. A perspective». Rapid Communications in Mass Spectrometry: e9354. doi:10.1002/rcm.9354. ISSN 0951-4198. PMID 35830299. S2CID 250491726.
- ^ Horning, E. C.; Horning, M. G.; Carroll, D. I.; Dzidic, I.; Stillwell, R. N. (1973-05-01). «New picogram detection system based on a mass spectrometer with an external ionization source at atmospheric pressure». Analytical Chemistry. 45 (6): 936–943. doi:10.1021/ac60328a035. ISSN 0003-2700.
- ^ Robb, null; Covey, null; Bruins, null (2000-08-01). «Atmospheric pressure photoionization: an ionization method for liquid chromatography-mass spectrometry». Analytical Chemistry. 72 (15): 3653–3659. doi:10.1021/ac0001636. ISSN 1520-6882. PMID 10952556.
- ^ Widmer, Leo; Watson, Stuart; Schlatter, Konrad; Crowson, Andrew (2002). «Development of an LC/MS method for the trace analysis of triacetone triperoxide (TATP)». The Analyst. 127 (12): 1627–1632. Bibcode:2002Ana…127.1627W. doi:10.1039/b208350g. ISSN 0003-2654. PMID 12537371.
- ^ a b Sudhakar, P.; Latha, P.; Reddy, P. V. (2016-04-05). Phenotyping Crop Plants for Physiological and Biochemical Traits. Academic Press. ISBN 9780128041109.
- ^ Wysocki VH, Resing KA, Zhang Q, Cheng G (2005). «Mass spectrometry of peptides and proteins». Methods. 35 (3): 211–22. doi:10.1016/j.ymeth.2004.08.013. PMID 15722218.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Karlsson, Roger; Thorsell, Annika; Gomila, Margarita; Salvà-Serra, Francisco; Jakobsson, Hedvig E.; Gonzales-Siles, Lucia; Jaén-Luchoro, Daniel; Skovbjerg, Susann; Fuchs, Johannes; Karlsson, Anders; Boulund, Fredrik (2020-03-01). «Discovery of Species-unique Peptide Biomarkers of Bacterial Pathogens by Tandem Mass Spectrometry-based Proteotyping». Molecular & Cellular Proteomics. 19 (3): 518–528. doi:10.1074/mcp.RA119.001667. ISSN 1535-9476. PMC 7050107. PMID 31941798.
- ^ Van Puyvelde, B; Van Uytfanghe, K; Tytgat, O; Van Oudenhove, L; Gabriels, R; Bouwmeester, R; Daled, S; Van Den Bossche, T; Ramasamy, P; Verhelst, S; De Clerck, L; Corveleyn, L; Willems, S; Debunne, N; Wynendaele, E; De Spiegeleer, B; Judak, P; Roels, K; De Wilde, L; Van Eenoo, P; Reyns, T; Cherlet, M; Dumont, E; Debyser, G; t’Kindt, R; Sandra, K; Gupta, S; Drouin, N; Harms, A; Hankemeier, T; Jones, DJL; Gupta, P; Lane, D; Lane, CS; El Ouadi, S; Vincendet, JB; Morrice, N; Oehrle, S; Tanna, N; Silvester, S; Hannam, S; Sigloch, FC; Bhangu-Uhlmann, A; Claereboudt, J; Anderson, NL; Razavi, M; Degroeve, S; Cuypers, L; Stove, C; Lagrou, K; Martens, GA; Deforce, D; Martens, L; Vissers, JPC; Dhaenens, M (28 June 2021). «Cov-MS: A Community-Based Template Assay for Mass-Spectrometry-Based Protein Detection in SARS-CoV-2 Patients». JACS Au. 1 (6): 750–765. doi:10.1021/jacsau.1c00048. PMC 8230961. PMID 34254058.
- ^ Gika, Helen G.; Theodoridis, Georgios A.; Plumb, Robert S.; Wilson, Ian D. (January 2014). «Current practice of liquid chromatography–mass spectrometry in metabolomics and metabonomics». Journal of Pharmaceutical and Biomedical Analysis. 87: 12–25. doi:10.1016/j.jpba.2013.06.032. ISSN 0731-7085. PMID 23916607.
- ^ Stobiecki, M.; Skirycz, A.; Kerhoas, L.; Kachlicki, P.; Muth, D.; Einhorn, J.; Mueller-Roeber, B. (2006). «Profiling of phenolic glycosidic conjugates in leaves of Arabidopsis thaliana using LC/MS». Metabolomics. 2 (4): 197–219. doi:10.1007/s11306-006-0031-5. S2CID 39140266.
- ^ Jorge, Tiago F.; Rodrigues, João A.; Caldana, Camila; Schmidt, Romy; van Dongen, Joost T.; Thomas-Oates, Jane; António, Carla (2016-09-01). «Mass spectrometry-based plant metabolomics: Metabolite responses to abiotic stress». Mass Spectrometry Reviews. 35 (5): 620–649. Bibcode:2016MSRv…35..620J. doi:10.1002/mas.21449. ISSN 1098-2787. PMID 25589422.
- ^ Tolstikov, Vladimir V.; Fiehn, Oliver (2002). «Analysis of Highly Polar Compounds of Plant Origin: Combination of Hydrophilic Interaction Chromatography and Electrospray Ion Trap Mass Spectrometry». Analytical Biochemistry. 301 (2): 298–307. doi:10.1006/abio.2001.5513. PMID 11814300. S2CID 3156968.
- ^ Antonio, Carla; Larson, Tony; Gilday, Alison; Graham, Ian; Bergström, Ed; Thomas-Oates, Jane (2008). «Hydrophilic interaction chromatography/electrospray mass spectrometry analysis of carbohydrate-related metabolites from Arabidopsis thaliana leaf tissue». Rapid Communications in Mass Spectrometry. 22 (9): 1399–1407. Bibcode:2008RCMS…22.1399A. doi:10.1002/rcm.3519. PMID 18384194.
- ^ Lee, Mike S.; Kerns, Edward H. (1999). «LC/MS applications in drug development». Mass Spectrometry Reviews. 18 (3–4): 187–279. Bibcode:1999MSRv…18..187L. doi:10.1002/(SICI)1098-2787(1999)18:3/4<187::AID-MAS2>3.0.CO;2-K. PMID 10568041.
Further reading[edit]
- Thurman, E. M.; Ferrer, Imma (2003). Liquid chromatography/mass spectrometry, MS/MS and time of flight MS: analysis of emerging contaminants. Columbus, OH: American Chemical Society. ISBN 978-0-8412-3825-1.
- Ferrer, Imma; Thurman, E. M. (2009). Liquid chromatography-Time of Flight Mass Spectrometry: Principles, Tools and Applications for Accurate Mass Analysis. New York, NJ: Wiley. ISBN 978-0-470-13797-0.
- McMaster, Marvin C. (2005). LC/MS: a practical user’s guide. New York: John Wiley. ISBN 978-0-471-65531-2.
- Yergey, Alfred L. (1990). Liquid chromatography/mass spectrometry: techniques and applications. New York: Plenum Press. ISBN 978-0-306-43186-9.