Муковисцидóз (кистозный фиброз) — системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза и характеризующееся поражением желёз внешней секреции, тяжёлыми нарушениями функций органов дыхания. Муковисцидоз представляет особый интерес не только из-за широкой распространённости, но и потому, что это одно из первых наследственных заболеваний, которое пытались лечить.
Все значения слова «муковисцидоз»
-
Мужчины и женщины с муковисцидозом доживают до зрелого возраста, о чём раньше не приходилось и мечтать; доноров почки ищут в Facebook; некоторые больные после проведения реанимационных мероприятий в отделении интенсивной терапии борются с посттравматическим стрессовым расстройством, другие оказываются до конца жизни прикованы к аппарату искусственной вентиляции лёгких в специализированных медицинских центрах.
-
Потому что защита больных муковисцидозом – это именно политика.
-
Европейское общество муковисцидоза уполномочило Carlo Castellani и Harry Heijerman собрать вместе коллектив высококвалифицированных специалистов в данной области для участия в этой монографии.
- (все предложения)
- лямблиоз
- аскаридоз
- кольпит
- гельминтоз
- лимфогранулематоз
- (ещё синонимы…)
| Cystic fibrosis | |
|---|---|
| Other names | Mucoviscidosis |
 |
|
| Specialty | Medical genetics, pulmonology |
| Symptoms | Difficulty breathing, coughing up mucus, poor growth, fatty stool[1] |
| Usual onset | Symptoms recognizable ~6 month[2] |
| Duration | Life long[3] |
| Causes | Genetic (autosomal recessive)[1] |
| Risk factors | Genetic |
| Diagnostic method | Sweat test, genetic testing[1] |
| Treatment | Antibiotics, pancreatic enzyme replacement, lung transplantation[1] |
| Prognosis | Life expectancy between 42 and 50 years (developed world)[4] |
| Frequency | 1 out of 3,000 (Northern European)[1] |
Cystic fibrosis (CF) is a rare[5][6] genetic disorder that affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine.[1][7] Long-term issues include difficulty breathing and coughing up mucus as a result of frequent lung infections.[1] Other signs and symptoms may include sinus infections, poor growth, fatty stool, clubbing of the fingers and toes, and infertility in most males.[1] Different people may have different degrees of symptoms.[1]
Cystic fibrosis is inherited in an autosomal recessive manner.[1] It is caused by the presence of mutations in both copies of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein.[1] Those with a single working copy are carriers and otherwise mostly healthy.[3] CFTR is involved in the production of sweat, digestive fluids, and mucus.[8] When the CFTR is not functional, secretions which are usually thin instead become thick.[9] The condition is diagnosed by a sweat test and genetic testing.[1] Screening of infants at birth takes place in some areas of the world.[1]
There is no known cure for cystic fibrosis.[3] Lung infections are treated with antibiotics which may be given intravenously, inhaled, or by mouth.[1] Sometimes, the antibiotic azithromycin is used long term.[1] Inhaled hypertonic saline and salbutamol may also be useful.[1] Lung transplantation may be an option if lung function continues to worsen.[1] Pancreatic enzyme replacement and fat-soluble vitamin supplementation are important, especially in the young.[1] Airway clearance techniques such as chest physiotherapy have some short-term benefit, but long-term effects are unclear.[10] The average life expectancy is between 42 and 50 years in the developed world.[4][11] Lung problems are responsible for death in 80% of people with cystic fibrosis.[1]
CF is most common among people of Northern European ancestry, for whom it affects about 1 out of 3,000 newborns,[1] and among which around 1 out of 25 people is a carrier.[3] It is least common in Africans and Asians, though it does occur in all races.[1] It was first recognized as a specific disease by Dorothy Andersen in 1938, with descriptions that fit the condition occurring at least as far back as 1595.[7] The name «cystic fibrosis» refers to the characteristic fibrosis and cysts that form within the pancreas.[7][12]
Signs and symptoms[edit]

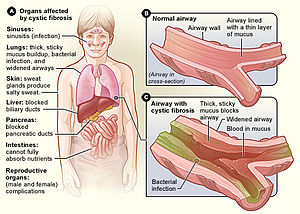
Health problems associated with cystic fibrosis
Cystic fibrosis typically manifests early in life. Newborns and infants with cystic fibrosis tend to have frequent, large, greasy stools (a result of malabsorption) and are underweight for their age.[13] 15–20% of newborns have their small intestine blocked by meconium, often requiring surgery to correct.[13] Newborns occasionally have neonatal jaundice due to blockage of the bile ducts.[13] Children with cystic fibrosis lose excessive salt in their sweat, and parents often notice salt crystallizing on the skin, or a salty taste when they kiss their child.[13]
The primary cause of morbidity and death in people with cystic fibrosis is progressive lung disease, which eventually leads to respiratory failure.[14] This typically begins as a prolonged respiratory infection that continues until treated with antibiotics.[14] Chronic infection of the respiratory tract is nearly universal in people with cystic fibrosis, with Pseudomonas aeruginosa, fungi, and mycobacteria all increasingly common over time.[15] Inflammation of the upper airway results in frequent runny nose and nasal obstruction. Nasal polyps are common, particularly in children and teenagers.[14] As the disease progresses, people tend to have shortness of breath, and a chronic cough that produces sputum.[14] Breathing problems make it increasingly challenging to exercise, and prolonged illness causes those affected to be underweight for their age.[14] In late adolescence or adulthood, people begin to develop severe signs of lung disease: wheezing, digital clubbing, cyanosis, coughing up blood, pulmonary heart disease, and collapsed lung (atelectasis or pneumothorax).[14]
In rare cases, cystic fibrosis can manifest itself as a coagulation disorder. Vitamin K is normally absorbed from breast milk, formula, and later, solid foods. This absorption is impaired in some CF patients. Young children are especially sensitive to vitamin K malabsorptive disorders because only a very small amount of vitamin K crosses the placenta, leaving the child with very low reserves and limited ability to absorb vitamin K from dietary sources after birth. Because clotting factors II, VII, IX, and X are vitamin K–dependent, low levels of vitamin K can result in coagulation problems. Consequently, when a child presents with unexplained bruising, a coagulation evaluation may be warranted to determine whether an underlying disease is present.[16]
Lungs and sinuses[edit]

Lung disease results from clogging of the airways due to mucus build-up, decreased mucociliary clearance, and resulting inflammation.[17][18] In later stages, changes in the architecture of the lung, such as pathology in the major airways (bronchiectasis), further exacerbate difficulties in breathing. Other signs include high blood pressure in the lung (pulmonary hypertension), heart failure, difficulties getting enough oxygen to the body (hypoxia), and respiratory failure requiring support with breathing masks, such as bilevel positive airway pressure machines or ventilators.[19] Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa are the three most common organisms causing lung infections in CF patients.[18]: 1254 In addition, opportunistic infection due to Burkholderia cepacia complex can occur, especially through transmission from patient to patient.[20]
In addition to typical bacterial infections, people with CF more commonly develop other types of lung diseases. Among these is allergic bronchopulmonary aspergillosis, in which the body’s response to the common fungus Aspergillus fumigatus causes worsening of breathing problems. Another is infection with Mycobacterium avium complex, a group of bacteria related to tuberculosis, which can cause lung damage and do not respond to common antibiotics.[21]
Mucus in the paranasal sinuses is equally thick and may also cause blockage of the sinus passages, leading to infection. This may cause facial pain, fever, nasal drainage, and headaches. Individuals with CF may develop overgrowth of the nasal tissue (nasal polyps) due to inflammation from chronic sinus infections.[22] Recurrent sinonasal polyps can occur in 10% to 25% of CF patients.[18]: 1254 These polyps can block the nasal passages and increase breathing difficulties.[23][24]
Cardiorespiratory complications are the most common causes of death (about 80%) in patients at most CF centers in the United States.[18]: 1254
Gastrointestinal[edit]
In addition, protrusion of internal rectal membranes (rectal prolapse) is more common, occurring in as many as 10% of children with CF,[18] and it is caused by increased fecal volume, malnutrition, and increased intra–abdominal pressure due to coughing.[25]
The thick mucus seen in the lungs has a counterpart in thickened secretions from the pancreas, an organ responsible for providing digestive juices that help break down food. These secretions block the exocrine movement of the digestive enzymes into the duodenum and result in irreversible damage to the pancreas, often with painful inflammation (pancreatitis).[26] The pancreatic ducts are totally plugged in more advanced cases, usually seen in older children or adolescents.[18] This causes atrophy of the exocrine glands and progressive fibrosis.[18]
Individuals with CF also have difficulties absorbing the fat-soluble vitamins A, D, E, and K.[27]
In addition to the pancreas problems, people with CF experience more heartburn,[27] intestinal blockage by intussusception, and constipation.[28] Older individuals with CF may develop distal intestinal obstruction syndrome, which occurs when feces becomes thick with mucus (inspissated) and can cause bloating, pain, and incomplete or complete bowel obstruction.[29][27]
Exocrine pancreatic insufficiency occurs in the majority (85–90%) of patients with CF.[18]: 1253 It is mainly associated with «severe» CFTR mutations, where both alleles are completely nonfunctional (e.g. ΔF508/ΔF508).[18]: 1253 It occurs in 10–15% of patients with one «severe» and one «mild» CFTR mutation where little CFTR activity still occurs, or where two «mild» CFTR mutations exist.[18]: 1253 In these milder cases, sufficient pancreatic exocrine function is still present so that enzyme supplementation is not required.[18]: 1253 Usually, no other GI complications occur in pancreas-sufficient phenotypes, and in general, such individuals usually have excellent growth and development.[18]: 1254 Despite this, idiopathic chronic pancreatitis can occur in a subset of pancreas-sufficient individuals with CF, and is associated with recurrent abdominal pain and life-threatening complications.[18]
Thickened secretions also may cause liver problems in patients with CF. Bile secreted by the liver to aid in digestion may block the bile ducts, leading to liver damage. Impaired digestion or absorption of lipids can result in steatorrhea. Over time, this can lead to scarring and nodularity (cirrhosis). The liver fails to rid the blood of toxins and does not make important proteins, such as those responsible for blood clotting.[30][31] Liver disease is the third-most common cause of death associated with CF.[18]
Around 5–7% of people experience liver damage severe enough to cause symptoms: typically gallstones causing biliary colic.[32]
Endocrine[edit]
The pancreas contains the islets of Langerhans, which are responsible for making insulin, a hormone that helps regulate blood glucose. Damage to the pancreas can lead to loss of the islet cells, leading to a type of diabetes unique to those with the disease.[33] This cystic fibrosis-related diabetes shares characteristics of type 1 and type 2 diabetes, and is one of the principal nonpulmonary complications of CF.[34]
Vitamin D is involved in calcium and phosphate regulation. Poor uptake of vitamin D from the diet because of malabsorption can lead to the bone disease osteoporosis in which weakened bones are more susceptible to fractures.[35]
Infertility[edit]
Infertility affects both men and women. At least 97% of men with cystic fibrosis are infertile, but not sterile, and can have children with assisted reproductive techniques.[36] The main cause of infertility in men with cystic fibrosis is congenital absence of the vas deferens (which normally connects the testes to the ejaculatory ducts of the penis), but potentially also by other mechanisms such as causing no sperm, abnormally shaped sperm, and few sperm with poor motility.[37] Many men found to have congenital absence of the vas deferens during evaluation for infertility have a mild, previously undiagnosed form of CF.[38] Around 20% of women with CF have fertility difficulties due to thickened cervical mucus or malnutrition. In severe cases, malnutrition disrupts ovulation and causes a lack of menstruation.[39]
Causes[edit]
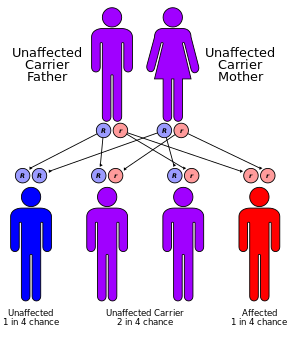
Cystic fibrosis has an autosomal recessive pattern of inheritance.
CF is caused by a mutation in the gene cystic fibrosis transmembrane conductance regulator (CFTR). The most common mutation, ΔF508, is a deletion (Δ signifying deletion) of three nucleotides that results in a loss of the amino acid phenylalanine (F) at the 508th position on the protein.[40][41] This mutation accounts for two-thirds (66–70%[18]) of CF cases worldwide and 90% of cases in the United States; however, over 1500 other mutations can produce CF.[42] Although most people have two working copies (alleles) of the CFTR gene, only one is needed to prevent cystic fibrosis. CF develops when neither allele can produce a functional CFTR protein. Thus, CF is considered an autosomal recessive disease.[43]
The CFTR gene, found at the q31.2 locus of chromosome 7, is 230,000 base pairs long, and creates a protein that is 1,480 amino acids long. More specifically, the location is between base pair 117,120,016 and 117,308,718 on the long arm of chromosome 7, region 3, band 1, subband 2, represented as 7q31.2. Structurally, the CFTR is a type of gene known as an ABC gene. The product of this gene (the CFTR protein) is a chloride ion channel important in creating sweat, digestive juices, and mucus. This protein possesses two ATP-hydrolyzing domains, which allows the protein to use energy in the form of ATP. It also contains two domains comprising six alpha helices apiece, which allow the protein to cross the cell membrane. A regulatory binding site on the protein allows activation by phosphorylation, mainly by cAMP-dependent protein kinase.[19] The carboxyl terminal of the protein is anchored to the cytoskeleton by a PDZ domain interaction.[44] The majority of CFTR in the lung’s passages is produced by rare ion-transporting cells that regulate mucus properties.[45]
In addition, the evidence is increasing that genetic modifiers besides CFTR modulate the frequency and severity of the disease. One example is mannan-binding lectin, which is involved in innate immunity by facilitating phagocytosis of microorganisms. Polymorphisms in one or both mannan-binding lectin alleles that result in lower circulating levels of the protein are associated with a threefold higher risk of end-stage lung disease, as well as an increased burden of chronic bacterial infections.[18]
Carriers[edit]
Up to one in 25 individuals of Northern European ancestry is considered a genetic carrier. The disease appears only when two of these carriers have children, as each pregnancy between them has a 25% chance of producing a child with the disease. Although only about one of every 3,000 newborns of the affected ancestry has CF, more than 900 mutations of the gene that causes CF are known. Current tests look for the most common mutations.[46]
The mutations screened by the test vary according to a person’s ethnic group or by the occurrence of CF already in the family. More than 10 million Americans, including one in 25 white Americans, are carriers of one mutation of the CF gene. CF is present in other races, though not as frequently as in white individuals. About one in 46 Hispanic Americans, one in 65 African Americans, and one in 90 Asian Americans carry a mutation of the CF gene.[46]
Pathophysiology[edit]

The CFTR protein is a channel protein that controls the flow of H2O and Cl− ions in and out of cells inside the lungs. When the CFTR protein is working correctly, ions freely flow in and out of the cells. However, when the CFTR protein is malfunctioning, these ions cannot flow out of the cell due to a blocked channel. This causes cystic fibrosis, characterized by the buildup of thick mucus in the lungs.
Several mutations in the CFTR gene can occur, and different mutations cause different defects in the CFTR protein, sometimes causing a milder or more severe disease. These protein defects are also targets for drugs which can sometimes restore their function. ΔF508-CFTR gene mutation, which occurs in >90% of patients in the U.S., creates a protein that does not fold normally and is not appropriately transported to the cell membrane, resulting in its degradation.[47]
Other mutations result in proteins that are too short (truncated) because production is ended prematurely. Other mutations produce proteins that do not use energy (in the form of ATP) normally, do not allow chloride, iodide, and thiocyanate to cross the membrane appropriately,[48] and degrade at a faster rate than normal. Mutations may also lead to fewer copies of the CFTR protein being produced.[19]
The protein created by this gene is anchored to the outer membrane of cells in the sweat glands, lungs, pancreas, and all other remaining exocrine glands in the body.
The protein spans this membrane and acts as a channel connecting the inner part of the cell (cytoplasm) to the surrounding fluid. This channel is primarily responsible for controlling the movement of halide anions from inside to outside of the cell; however, in the sweat ducts, it facilitates the movement of chloride from the sweat duct into the cytoplasm. When the CFTR protein does not resorb ions in sweat ducts, chloride and thiocyanate[49] released from sweat glands are trapped inside the ducts and pumped to the skin.
Additionally hypothiocyanite, OSCN, cannot be produced by the immune defense system.[50][51] Because chloride is negatively charged, this modifies the electrical potential inside and outside the cell that normally causes cations to cross into the cell. Sodium is the most common cation in the extracellular space. The excess chloride within sweat ducts prevents sodium resorption by epithelial sodium channels and the combination of sodium and chloride creates the salt, which is lost in high amounts in the sweat of individuals with CF. This lost salt forms the basis for the sweat test.[19]
Most of the damage in CF is due to blockage of the narrow passages of affected organs with thickened secretions. These blockages lead to remodeling and infection in the lung, damage by accumulated digestive enzymes in the pancreas, blockage of the intestines by thick feces, etc. Several theories have been posited on how the defects in the protein and cellular function cause the clinical effects. The most current theory suggests that defective ion transport leads to dehydration in the airway epithelia, thickening mucus.[52] In airway epithelial cells, the cilia exist in between the cell’s apical surface and mucus in a layer known as airway surface liquid (ASL). The flow of ions from the cell and into this layer is determined by ion channels such as CFTR. CFTR not only allows chloride ions to be drawn from the cell and into the ASL, but it also regulates another channel called ENac, which allows sodium ions to leave the ASL and enter the respiratory epithelium. CFTR normally inhibits this channel, but if the CFTR is defective, then sodium flows freely from the ASL and into the cell.[citation needed]
As water follows sodium, the depth of ASL will be depleted and the cilia will be left in the mucous layer.[53] As cilia cannot effectively move in a thick, viscous environment, mucociliary clearance is deficient and a buildup of mucus occurs, clogging small airways.[54] The accumulation of more viscous, nutrient-rich mucus in the lungs allows bacteria to hide from the body’s immune system, causing repeated respiratory infections. The presence of the same CFTR proteins in the pancreatic duct and sweat glands in the skin also cause symptoms in these systems.[citation needed]
Chronic infections[edit]
The lungs of individuals with cystic fibrosis are colonized and infected by bacteria from an early age. These bacteria, which often spread among individuals with CF, thrive in the altered mucus, which collects in the small airways of the lungs. This mucus leads to the formation of bacterial microenvironments known as biofilms that are difficult for immune cells and antibiotics to penetrate. Viscous secretions and persistent respiratory infections repeatedly damage the lung by gradually remodeling the airways, which makes infection even more difficult to eradicate.[55] The natural history of CF lung infections and airway remodeling is poorly understood, largely due to the immense spatial and temporal heterogeneity both within and between the microbiomes of CF patients.[56]
Over time, both the types of bacteria and their individual characteristics change in individuals with CF. In the initial stage, common bacteria such as S. aureus and H. influenzae colonize and infect the lungs.[18] Eventually, Pseudomonas aeruginosa (and sometimes Burkholderia cepacia) dominates. By 18 years of age, 80% of patients with classic CF harbor P. aeruginosa, and 3.5% harbor B. cepacia.[18] Once within the lungs, these bacteria adapt to the environment and develop resistance to commonly used antibiotics. Pseudomonas can develop special characteristics that allow the formation of large colonies, known as «mucoid» Pseudomonas, which are rarely seen in people who do not have CF.[55] Scientific evidence suggests the interleukin 17 pathway plays a key role in resistance and modulation of the inflammatory response during P. aeruginosa infection in CF.[57] In particular, interleukin 17-mediated immunity plays a double-edged activity during chronic airways infection; on one side, it contributes to the control of P. aeruginosa burden, while on the other, it propagates exacerbated pulmonary neutrophilia and tissue remodeling.[57]
Infection can spread by passing between different individuals with CF.[58] In the past, people with CF often participated in summer «CF camps» and other recreational gatherings.[59][60] Hospitals grouped patients with CF into common areas and routine equipment (such as nebulizers)[61] was not sterilized between individual patients.[62] This led to transmission of more dangerous strains of bacteria among groups of patients. As a result, individuals with CF are now routinely isolated from one another in the healthcare setting, and healthcare providers are encouraged to wear gowns and gloves when examining patients with CF to limit the spread of virulent bacterial strains.[63]
CF patients may also have their airways chronically colonized by filamentous fungi (such as Aspergillus fumigatus, Scedosporium apiospermum, Aspergillus terreus) and/or yeasts (such as Candida albicans); other filamentous fungi less commonly isolated include Aspergillus flavus and Aspergillus nidulans (occur transiently in CF respiratory secretions) and Exophiala dermatitidis and Scedosporium prolificans (chronic airway-colonizers); some filamentous fungi such as Penicillium emersonii and Acrophialophora fusispora are encountered in patients almost exclusively in the context of CF.[64] Defective mucociliary clearance characterizing CF is associated with local immunological disorders. In addition, the prolonged therapy with antibiotics and the use of corticosteroid treatments may also facilitate fungal growth. Although the clinical relevance of the fungal airway colonization is still a matter of debate, filamentous fungi may contribute to the local inflammatory response and therefore to the progressive deterioration of the lung function, as often happens with allergic bronchopulmonary aspergillosis – the most common fungal disease in the context of CF, involving a Th2-driven immune response to Aspergillus species.[64][65]
Diagnosis[edit]

The location of the CFTR gene on chromosome 7
In many localities all newborns are screened for cystic fibrosis within the first few days of life, typically by blood test for high levels of immunoreactive trypsinogen.[66] Newborns with positive tests or those who are otherwise suspected of having cystic fibrosis based on symptoms or family history, then undergo a sweat test. An electric current is used to drive pilocarpine into the skin, stimulating sweating. The sweat is collected and analyzed for salt levels. Having unusually high levels of chloride in the sweat suggests CFTR is dysfunctional; the person is then diagnosed with cystic fibrosis.[67][note 1] Genetic testing is also available to identify the CFTR mutations typically associated with cystic fibrosis. Many laboratories can test for the 30–96 most common CFTR mutations, which can identify over 90% of people with cystic fibrosis.[67]
People with CF have less thiocyanate and hypothiocyanite in their saliva[69] and mucus (Banfi et al.). In the case of milder forms of CF, transepithelial potential difference measurements can be helpful. CF can also be diagnosed by identification of mutations in the CFTR gene.[70]
In many cases, a parent makes the diagnosis because the infant tastes salty.[18] Immunoreactive trypsinogen levels can be increased in individuals who have a single mutated copy of the CFTR gene (carriers) or, in rare instances, in individuals with two normal copies of the CFTR gene. Due to these false positives, CF screening in newborns can be controversial.[71][72]
By 2010 every US state had instituted newborn screening programs [73] and as of 2016, 21 European countries had programs in at least some regions.[74]
Prenatal[edit]
Women who are pregnant or couples planning a pregnancy can have themselves tested for the CFTR gene mutations to determine the risk that their child will be born with CF. Testing is typically performed first on one or both parents and, if the risk of CF is high, testing on the fetus is performed. The American College of Obstetricians and Gynecologists recommends all people thinking of becoming pregnant be tested to see if they are a carrier.[75]
Because development of CF in the fetus requires each parent to pass on a mutated copy of the CFTR gene and because CF testing is expensive, testing is often performed initially on one parent. If testing shows that parent is a CFTR gene mutation carrier, the other parent is tested to calculate the risk that their children will have CF. CF can result from more than a thousand different mutations.[43] As of 2016, typically only the most common mutations are tested for, such as ΔF508[43] Most commercially available tests look for 32 or fewer different mutations. If a family has a known uncommon mutation, specific screening for that mutation can be performed. Because not all known mutations are found on current tests, a negative screen does not guarantee that a child will not have CF.[76]
During pregnancy, testing can be performed on the placenta (chorionic villus sampling) or the fluid around the fetus (amniocentesis). However, chorionic villus sampling has a risk of fetal death of one in 100 and amniocentesis of one in 200;[77] a recent study has indicated this may be much lower, about one in 1,600.[78]
Economically, for carrier couples of cystic fibrosis, when comparing preimplantation genetic diagnosis (PGD) with natural conception (NC) followed by prenatal testing and abortion of affected pregnancies, PGD provides net economic benefits up to a maternal age around 40 years, after which NC, prenatal testing, and abortion have higher economic benefit.[79]
Management[edit]
While no cures for CF are known, several treatment methods are used. The management of CF has improved significantly over the past 70 years. While infants born with it 70 years ago would have been unlikely to live beyond their first year, infants today are likely to live well into adulthood. Recent advances in the treatment of cystic fibrosis have meant that individuals with cystic fibrosis can live a fuller life less encumbered by their condition. The cornerstones of management are the proactive treatment of airway infection, and encouragement of good nutrition and an active lifestyle. Pulmonary rehabilitation as a management of CF continues throughout a person’s life, and is aimed at maximizing organ function, and therefore the quality of life. Occupational therapists use energy conservation techniques (ECT) in the rehabilitation process for patients with Cystic Fibrosis.[80] Examples of energy conservation techniques are ergonomic principles, pursed lip breathing, and diaphragmatic breathing.[81] Patients with CF tend to have fatigue and dyspnoea due to chronic pulmonary infections, so reducing the amount of energy spent during activities can help patients feel better and gain more independence.[80] At best, current treatments delay the decline in organ function. Because of the wide variation in disease symptoms, treatment typically occurs at specialist multidisciplinary centers and is tailored to the individual. Targets for therapy are the lungs, gastrointestinal tract (including pancreatic enzyme supplements), the reproductive organs (including assisted reproductive technology), and psychological support.[82]
The most consistent aspect of therapy in CF is limiting and treating the lung damage caused by thick mucus and infection, with the goal of maintaining quality of life. Intravenous, inhaled, and oral antibiotics are used to treat chronic and acute infections. Mechanical devices and inhalation medications are used to alter and clear the thickened mucus. These therapies, while effective, can be extremely time-consuming. Oxygen therapy at home is recommended in those with significant low oxygen levels.[83] Many people with CF use probiotics, which are thought to be able to correct intestinal dysbiosis and inflammation, but the clinical trial evidence regarding the effectiveness of probiotics for reducing pulmonary exacerbations in people with CF is uncertain.[84]
Antibiotics[edit]
Many people with CF are on one or more antibiotics at all times, even when healthy, to prophylactically suppress infection. Antibiotics are absolutely necessary whenever pneumonia is suspected or a noticeable decline in lung function is seen, and are usually chosen based on the results of a sputum analysis and the person’s past response. This prolonged therapy often necessitates hospitalization and insertion of a more permanent IV such as a peripherally inserted central catheter or Port-a-Cath. Inhaled therapy with antibiotics such as tobramycin, colistin, and aztreonam is often given for months at a time to improve lung function by impeding the growth of colonized bacteria.[85][86][87] Inhaled antibiotic therapy helps lung function by fighting infection, but also has significant drawbacks such as development of antibiotic resistance, tinnitus, and changes in the voice.[88] Inhaled levofloxacin may be used to treat Pseudomonas aeruginosa in people with cystic fibrosis who are infected.[89] The early management of Pseudomonas aeruginosa infection is easier and better, using nebulised antibiotics with or without oral antibiotics may sustain its eradication up to two years.[90] When choosing antibiotics to treat CF patients with lung infections caused by Pseudomonas aeruginosa in people with cystic fibrosis, it is still unclear whether the choice of antibiotics should be based on the results of testing antibiotics separately (one at a time) or in combination with each other.[91]
Antibiotics by mouth such as ciprofloxacin or azithromycin are given to help prevent infection or to control ongoing infection.[92] The aminoglycoside antibiotics (e.g. tobramycin) used can cause hearing loss, damage to the balance system in the inner ear or kidney failure with long-term use.[93] To prevent these side-effects, the amount of antibiotics in the blood is routinely measured and adjusted accordingly.[94]
All these factors related to the antibiotics use, the chronicity of the disease, and the emergence of resistant bacteria demand more exploration for different strategies such as antibiotic adjuvant therapy.[95] Currently, no reliable clinical trial evidence shows the effectiveness of antibiotics for pulmonary exacerbations in people with cystic fibrosis and Burkholderia cepacia complex[96] or for the use of antibiotics to treat nontuberculous mycobacteria in people with CF.[97]
Other medication[edit]
Aerosolized medications that help loosen secretions include dornase alfa and hypertonic saline.[98] Dornase is a recombinant human deoxyribonuclease, which breaks down DNA in the sputum, thus decreasing its viscosity.[99] Dornase alpha improves lung function and probably decreases the risk of exacerbations but there is insufficient evidence to know if it is more or less effective than other similar medications.[100] Dornase alpha may improve lung function, however there is no strong evidence that it is better than other hyperosmolar therapies.[100]
Denufosol, an investigational drug, opens an alternative chloride channel, helping to liquefy mucus.[101] Whether inhaled corticosteroids are useful is unclear, but stopping inhaled corticosteroid therapy is safe.[102] There is weak evidence that corticosteroid treatment may cause harm by interfering with growth.[102] Pneumococcal vaccination has not been studied as of 2014.[103] As of 2014, there is no clear evidence from randomized controlled trials that the influenza vaccine is beneficial for people with cystic fibrosis.[104]
Ivacaftor is a medication taken by mouth for the treatment of CF due to a number of specific mutations responsive to ivacaftor-induced CFTR protein enhancement.[105][106] It improves lung function by about 10%; however, as of 2014 it is expensive.[105] The first year it was on the market, the list price was over $300,000 per year in the United States.[105][needs update] In July 2015, the U.S. Food and Drug Administration approved lumacaftor/ivacaftor.[107] In 2018, the FDA approved the combination ivacaftor/tezacaftor; the manufacturer announced a list price of $292,000 per year.[108] Tezacaftor helps move the CFTR protein to the correct position on the cell surface, and is designed to treat people with the F508del mutation.[109]
In 2019, the combination drug elexacaftor/ivacaftor/tezacaftor marketed as Trikafta in the United States, was approved for CF patients over the age of 12.[110][111] In 2021, this was extended to include patients over the age of 6.[112] In Europe this drug was approved in 2020 and marketed as Kaftrio.[113] It is used in those that have a f508del mutation, which occurs in about 90% of patients with cystic fibrosis.[110][114] According to the Cystic Fibrosis Foundation, «this medicine represents the single greatest therapeutic advancement in the history of CF, offering a treatment for the underlying cause of the disease that could eventually bring modulator therapy to 90 percent of people with CF.»[115] In a clinical trial, participants who were administered the combination drug experienced a subsequent 63% decrease in pulmonary exacerbations and a 41.8 mmol/L decrease in sweat chloride concentration.[116] By mitigating a repertoire of symptoms associated with cystic fibrosis, the combination drug significantly improved quality-of-life metrics among patients with the disease as well.[116][115] The combination drug is also known to interact with CYP3A inducers, such as carbamazepine used in the treatment of bipolar disorder, causing elexafaftor/ivacaftor/tezacaftor to circulate in the body at decreased concentrations. As such, concomitant use is not recommended.[117] The list price in the US is going to be $311,000 per year;[118] however, insurance may cover much of the cost of the drug.[119]
Ursodeoxycholic acid, a bile salt, has been used, however there is insufficient data to show if it is effective.[120]
Nutrient supplementation[edit]
It is uncertain whether vitamin A or beta-carotene supplementation have any effect on eye and skin problems caused by vitamin A deficiency.[121]
There is no strong evidence that people with cystic fibrosis can prevent osteoporosis by increasing their intake of vitamin D.[122]
For people with vitamin E deficiency and cystic fibrosis, there is evidence that vitamin E supplementation may improve vitamin E levels, although it is still uncertain what effect supplementation has on vitamin E‐specific deficiency disorders or on lung function.[123]
Robust evidence regarding the effects of vitamin K supplementation in people with cystic fibrosis is lacking as of 2020.[124]
Various studies have examined the effects of omega-3 fatty acid supplementation for people with cystic fibrosis but the evidence is uncertain whether it has any benefits or adverse effects.[125]
Procedures[edit]
Several mechanical techniques are used to dislodge sputum and encourage its expectoration. One technique good for short-term airway clearance is chest physiotherapy where a respiratory therapist percusses an individual’s chest by hand several times a day, to loosen up secretions. This «percussive effect» can be administered also through specific devices that use chest wall oscillation or intrapulmonary percussive ventilator. Other methods such as biphasic cuirass ventilation, and associated clearance mode available in such devices, integrate a cough assistance phase, as well as a vibration phase for dislodging secretions. These are portable and adapted for home use.[10]
Another technique is positive expiratory pressure physiotherapy that consists of providing a back pressure to the airways during expiration. This effect is provided by devices that consists of a mask or a mouthpiece in which a resistance is applied only on the expiration phase.[126] Operating principles of this technique seems to be the increase of gas pressure behind mucus through collateral ventilation along with a temporary increase in functional residual capacity preventing the early collapse of small airways during exhalation.[127][128]
As lung disease worsens, mechanical breathing support may become necessary. Individuals with CF may need to wear special masks at night to help push air into their lungs. These machines, known as bilevel positive airway pressure (BiPAP) ventilators, help prevent low blood oxygen levels during sleep. Non-invasive ventilators may be used during physical therapy to improve sputum clearance.[129] It is not known if this type of therapy has an impact on pulmonary exacerbations or disease progression.[129] It is not known what role non-invasive ventilation therapy has for improving exercise capacity in people with cystic fibrosis.[129] However, the authors noted that «non‐invasive ventilation may be a useful adjunct to other airway clearance techniques, particularly in people with cystic fibrosis who have difficulty expectorating sputum.»[130] During severe illness, a tube may be placed in the throat (a procedure known as a tracheostomy) to enable breathing supported by a ventilator.[131][132]
For children, preliminary studies show massage therapy may help people and their families’ quality of life.[133]
Some lung infections require surgical removal of the infected part of the lung. If this is necessary many times, lung function is severely reduced.[134] The most effective treatment options for people with CF who have spontaneous or recurrent pneumothoraces is not clear.[135]
Transplantation[edit]
Lung transplantation may become necessary for individuals with CF as lung function and exercise tolerance decline. Although single lung transplantation is possible in other diseases, individuals with CF must have both lungs replaced because the remaining lung might contain bacteria that could infect the transplanted lung. A pancreatic or liver transplant may be performed at the same time to alleviate liver disease and/or diabetes.[136] Lung transplantation is considered when lung function declines to the point where assistance from mechanical devices is required or someone’s survival is threatened.[137] According to Merck Manual, «bilateral lung transplantation for severe lung disease is becoming more routine and more successful with experience and improved techniques. Among adults with CF, median survival posttransplant is about 9 years.»[138]
Other aspects[edit]

Intracytoplasmic sperm injection can be used to provide fertility for men with cystic fibrosis.
Newborns with intestinal obstruction typically require surgery, whereas adults with distal intestinal obstruction syndrome typically do not. Treatment of pancreatic insufficiency by replacement of missing digestive enzymes allows the duodenum to properly absorb nutrients and vitamins that would otherwise be lost in the feces. However, the best dosage and form of pancreatic enzyme replacement is unclear, as are the risks and long-term effectiveness of this treatment.[139]
So far, no large-scale research involving the incidence of atherosclerosis and coronary heart disease in adults with cystic fibrosis has been conducted. This is likely because the vast majority of people with cystic fibrosis do not live long enough to develop clinically significant atherosclerosis or coronary heart disease.[140]
Diabetes is the most common nonpulmonary complication of CF. It mixes features of type 1 and type 2 diabetes, and is recognized as a distinct entity, cystic fibrosis-related diabetes.[34][141] While oral antidiabetic drugs are sometimes used, the recommended treatment is the use of insulin injections or an insulin pump,[142] and, unlike in type 1 and 2 diabetes, dietary restrictions are not recommended.[34] While Stenotrophomonas maltophilia is relatively common in people with cystic fibrosis, the evidence about the effectiveness of antibiotics for S. maltophilia is uncertain.[143]
Bisphosphonates taken by mouth or intravenously can be used to improve the bone mineral density in people with cystic fibrosis.[144][needs update] When taking bisphosphates intravenously, adverse effects such as pain and flu-like symptoms can be an issue.[144] The adverse effects of bisphosphates taken by mouth on the gastrointestinal tract are not known.[144]
Poor growth may be avoided by insertion of a feeding tube for increasing food energy through supplemental feeds or by administration of injected growth hormone.[145]
Sinus infections are treated by prolonged courses of antibiotics. The development of nasal polyps or other chronic changes within the nasal passages may severely limit airflow through the nose, and over time reduce the person’s sense of smell. Sinus surgery is often used to alleviate nasal obstruction and to limit further infections. Nasal steroids such as fluticasone propionate are used to decrease nasal inflammation.[146]
Female infertility may be overcome by assisted reproduction technology, particularly embryo transfer techniques. Male infertility caused by absence of the vas deferens may be overcome with testicular sperm extraction, collecting sperm cells directly from the testicles. If the collected sample contains too few sperm cells to likely have a spontaneous fertilization, intracytoplasmic sperm injection can be performed.[147] Third party reproduction is also a possibility for women with CF. Whether taking antioxidants affects outcomes is unclear.[148]
Physical exercise is usually part of outpatient care for people with cystic fibrosis.[149] Aerobic exercise seems to be beneficial for aerobic exercise capacity, lung function and health-related quality of life; however, the quality of the evidence was poor.[149]
Due to the use of aminoglycoside antibiotics, ototoxicity is common. Symptoms may include «tinnitus, hearing loss, hyperacusis, aural fullness, dizziness, and vertigo».[150]
Gastrointestinal[edit]
Problems with the gastrointestinal system including constipation and obstruction of the gastrointestinal tract including distal intestinal obstruction syndrome are frequent complications for people with cystic fibrosis.[29] Treatment of gastrointestinal problems is required in order to prevent a complete obstruction, reduce other CF symptoms, and improve the quality of life.[29] While stool softeners, laxatives, and prokinetics (GI-focused treatments) are often suggested, there is no clear consensus from experts at to which approach is the best and comes with the least risks.[29] Mucolytics or systemic treatments aimed at dysfunctional CFTR are also sometimes suggested to improve symptoms.[151]
Prognosis[edit]
The prognosis for cystic fibrosis has improved due to earlier diagnosis through screening and better treatment and access to health care. In 1959, the median age of survival of children with CF in the United States was six months.[152]
In 2010, survival is estimated to be 37 years for women and 40 for men.[153] In Canada, median survival increased from 24 years in 1982 to 47.7 in 2007.[154] In the United States those born with CF in 2016 have an predicted life expectancy of 47.7 when cared for in specialty clinics.[155]
In the US, of those with CF who are more than 18 years old as of 2009, 92% had graduated from high school, 67% had at least some college education, 15% were disabled, 9% were unemployed, 56% were single, and 39% were married or living with a partner.[156]
Quality of life[edit]
Chronic illnesses can be difficult to manage. CF is a chronic illness that affects the «digestive and respiratory tracts resulting in generalized malnutrition and chronic respiratory infections».[157] The thick secretions clog the airways in the lungs, which often cause inflammation and severe lung infections.[158][159] If it is compromised, it affects the quality of life of someone with CF and their ability to complete such tasks as everyday chores.[citation needed]
According to Schmitz and Goldbeck (2006), CF significantly increases emotional stress on both the individual and the family, «and the necessary time-consuming daily treatment routine may have further negative effects on quality of life».[160] However, Havermans and colleagues (2006) have established that young outpatients with CF who have participated in the Cystic Fibrosis Questionnaire-Revised «rated some quality of life domains higher than did their parents».[161] Consequently, outpatients with CF have a more positive outlook for themselves. As Merck Manual notes, «with appropriate support, most patients can make an age-appropriate adjustment at home and school. Despite myriad problems, the educational, occupational, and marital successes of patients are impressive.»[138]
Furthermore, there are many ways to enhance the quality of life in CF patients. Exercise is promoted to increase lung function. Integrating an exercise regimen into the CF patient’s daily routine can significantly improve quality of life.[162] No definitive cure for CF is known, but diverse medications are used, such as mucolytics, bronchodilators, steroids, and antibiotics, that have the purpose of loosening mucus, expanding airways, decreasing inflammation, and fighting lung infections, respectively.[163]
Epidemiology[edit]
| Mutation | Frequency worldwide[164] |
|---|---|
| ΔF508 | 66–70%[18] |
| G542X | 2.4% |
| G551D | 1.6% |
| N1303K | 1.3% |
| W1282X | 1.2% |
| All others | 27.5% |
Cystic fibrosis is the most common life-limiting autosomal recessive disease among people of European heritage.[165] In the United States, about 30,000 individuals have CF; most are diagnosed by six months of age. In Canada, about 4,000 people have CF.[166] Around 1 in 25 people of European descent, and one in 30 of white Americans,[167] is a carrier of a CF mutation. Although CF is less common in these groups, roughly one in 46 Hispanics, one in 65 Africans, and one in 90 Asians carry at least one abnormal CFTR gene.[168][169] Ireland has the world’s highest prevalence of CF, at one in 1353.[170]
Although technically a rare disease, CF is ranked as one of the most widespread life-shortening genetic diseases. It is most common among nations in the Western world. An exception is Finland, where only one in 80 people carries a CF mutation.[171] The World Health Organization states, «In the European Union, one in 2000–3000 newborns is found to be affected by CF».[172] In the United States, one in 3,500 children is born with CF.[173] In 1997, about one in 3,300 white children in the United States was born with CF. In contrast, only one in 15,000 African American children have it, and in Asian Americans, the rate was even lower at one in 32,000.[174]
Cystic fibrosis is diagnosed equally in males and females. For reasons that remain unclear, data have shown that males tend to have a longer life expectancy than females,[175][176] though recent studies suggest this gender gap may no longer exist, perhaps due to improvements in health care facilities.[177][178] A recent study from Ireland identified a link between the female hormone estrogen and worse outcomes in CF.[179]
The distribution of CF alleles varies among populations. The frequency of ΔF508 carriers has been estimated at one in 200 in northern Sweden, one in 143 in Lithuanians, and one in 38 in Denmark. No ΔF508 carriers were found among 171 Finns and 151 Saami people.[180] ΔF508 does occur in Finland, but it is a minority allele there. CF is known to occur in only 20 families (pedigrees) in Finland.[181]
Evolution[edit]
The ΔF508 mutation is estimated to be up to 52,000 years old.[182] Numerous hypotheses have been advanced as to why such a lethal mutation has persisted and spread in the human population. Other common autosomal recessive diseases such as sickle-cell anemia have been found to protect carriers from other diseases, an evolutionary trade-off known as heterozygote advantage. Resistance to the following have all been proposed as possible sources of heterozygote advantage:
- Cholera: With the discovery that cholera toxin requires normal host CFTR proteins to function properly, it was hypothesized that carriers of mutant CFTR genes benefited from resistance to cholera and other causes of diarrhea.[183][184] Further studies have not confirmed this hypothesis.[185][186]
- Typhoid: Normal CFTR proteins are also essential for the entry of Salmonella Typhi into cells,[187] suggesting that carriers of mutant CFTR genes might be resistant to typhoid fever. No in vivo study has yet confirmed this. In both cases, the low level of cystic fibrosis outside of Europe, in places where both cholera and typhoid fever are endemic, is not immediately explicable.
- Diarrhea: The prevalence of CF in Europe might be connected with the development of cattle domestication. In this hypothesis, carriers of a single mutant CFTR had some protection from diarrhea caused by lactose intolerance, before the mutations that created lactose tolerance appeared.[188]
- Tuberculosis: Another possible explanation is that carriers of the gene could have some resistance to tuberculosis.[189][190] This hypothesis is based on the thesis that CFTR gene mutation carriers have insufficient action in one of their enzymes – arylsulphatase — which is necessary for Mycobacterium tuberculosis virulence. As M. tuberculosis would use its host’s sources to affect the individual, and due to the lack of enzyme it could not presents its virulence, being a carrier of CFTR mutation could provide resistance against tuberculosis.[191]
History[edit]

CF is supposed to have appeared about 3,000 BC because of migration of peoples, gene mutations, and new conditions in nourishment.[192] Although the entire clinical spectrum of CF was not recognized until the 1930s, certain aspects of CF were identified much earlier. Indeed, literature from Germany and Switzerland in the 18th century warned «Wehe dem Kind, das beim Kuß auf die Stirn salzig schmeckt, es ist verhext und muss bald sterben» («Woe to the child who tastes salty from a kiss on the brow, for he is cursed and soon must die»), recognizing the association between the salt loss in CF and illness.[192]
In the 19th century, Carl von Rokitansky described a case of fetal death with meconium peritonitis, a complication of meconium ileus associated with CF. Meconium ileus was first described in 1905 by Karl Landsteiner.[192] In 1936, Guido Fanconi described a connection between celiac disease, cystic fibrosis of the pancreas, and bronchiectasis.[193]
In 1938, Dorothy Hansine Andersen published an article, «Cystic Fibrosis of the Pancreas and Its Relation to Celiac Disease: a Clinical and Pathological Study», in the American Journal of Diseases of Children. She was the first to describe the characteristic cystic fibrosis of the pancreas and to correlate it with the lung and intestinal disease prominent in CF.[12] She also first hypothesized that CF was a recessive disease and first used pancreatic enzyme replacement to treat affected children. In 1952, Paul di Sant’Agnese discovered abnormalities in sweat electrolytes; a sweat test was developed and improved over the next decade.[194]
The first linkage between CF and another marker (paraoxonase) was found in 1985 by Hans Eiberg, indicating that only one locus exists for CF. In 1988, the first mutation for CF, ΔF508, was discovered by Francis Collins, Lap-Chee Tsui, and John R. Riordan on the seventh chromosome. Subsequent research has found over 1,000 different mutations that cause CF.[citation needed]
Because mutations in the CFTR gene are typically small, classical genetics techniques had been unable to accurately pinpoint the mutated gene.[195] Using protein markers, gene-linkage studies were able to map the mutation to chromosome 7. Chromosome walking and chromosome jumping techniques were then used to identify and sequence the gene.[196] In 1989, Lap-Chee Tsui led a team of researchers at the Hospital for Sick Children in Toronto that discovered the gene responsible for CF. CF represents a classic example of how a human genetic disorder was elucidated strictly by the process of forward genetics.[citation needed]
Research[edit]
People with CF may be listed in a disease registry that allows researchers and doctors to track health results and identify candidates for clinical trials.[197]
Gene therapy[edit]
Gene therapy has been explored as a potential cure for CF. Results from clinical trials have shown limited success as of 2016, and using gene therapy as routine therapy is not suggested.[198] A small study published in 2015 found a small benefit.[199]
The focus of much CF gene therapy research is aimed at trying to place a normal copy of the CFTR gene into affected cells. Transferring the normal CFTR gene into the affected epithelium cells would result in the production of functional CFTR protein in all target cells, without adverse reactions or an inflammation response. To prevent the lung manifestations of CF, only 5–10% the normal amount of CFTR gene expression is needed.[200] Multiple approaches have been tested for gene transfer, such as liposomes and viral vectors in animal models and clinical trials. However, both methods were found to be relatively inefficient treatment options,[201] mainly because very few cells take up the vector and express the gene, so the treatment has little effect. Additionally, problems have been noted in cDNA recombination, such that the gene introduced by the treatment is rendered unusable.[202] There has been a functional repair in culture of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients.[203]
Phage therapy[edit]
Phage therapy is being studied for multidrug resistant bacteria in people with CF.[204][205]
Gene modulators[edit]
A number of small molecules that aim at compensating various mutations of the CFTR gene are under development. CFTR modulator therapies have been used in place of other types of genetic therapies. These therapies focus on the expression of a genetic mutation instead of the mutated gene itself. Modulators are split into two classes: potentiators and correctors. Potentiators act on the CFTR ion channels that are embedded in the cell membrane, and these types of drugs help open up the channel to allow transmembrane flow. Correctors are meant to assist in the transportation of nascent proteins, a protein that is formed by ribosomes before it is morphed into a specific shape, to the cell surface to be implemented into the cell membrane.[206]
Most target the transcription stage of genetic expression. One approach has been to try and develop medication that get the ribosome to overcome the stop codon and produce a full-length CFTR protein. About 10% of CF results from a premature stop codon in the DNA, leading to early termination of protein synthesis and truncated proteins. These drugs target nonsense mutations such as G542X, which consists of the amino acid glycine in position 542 being replaced by a stop codon. Aminoglycoside antibiotics interfere with protein synthesis and error-correction. In some cases, they can cause the cell to overcome a premature stop codon by inserting a random amino acid, thereby allowing expression of a full-length protein. Future research for these modulators is focused on the cellular targets that can be effected by a change in a gene’s expression. Otherwise, genetic therapy will be used as a treatment when modulator therapies do not work given that 10% of people with cystic fibrosis are not affected by these drugs.[207]
Elexacaftor/ivacaftor/tezacaftor was approved in the United States in 2019 for cystic fibrosis.[208] This combination of previously developed medicines is able to treat up to 90% of people with cystic fibrosis.[206][208] This medications restores some effectiveness of the CFTR protein so that it can work as an ion channel on the cell’s surface.[209]
Ecological therapy[edit]
It has previously been shown that inter-species interactions are an important contributor to the pathology of CF lung infections. Examples include the production of antibiotic degrading enzymes such as β-lactamases and the production of metabolic by-products such as short-chain fatty acids (SCFAs) by anaerobic species, which can enhance the pathogenicity of traditional pathogens such as Pseudomonas aeruginosa.[210] Due to this, it has been suggested that the direct alteration of CF microbial community composition and metabolic function would provide an alternative to traditional antibiotic therapies.[56]
Society and culture[edit]
- Salt in My Soul: An Unfinished Life a posthumous memoir by Mallory Smith, a Californian with CF
- Sick: The Life and Death of Bob Flanagan, Supermasochist, a 1997 documentary film
- 65 Redroses, a 2009 documentary film
- Breathing for a Living, a memoir by Laura Rothenberg
- Every Breath I Take, Surviving and Thriving With Cystic Fibrosis, book by Claire Wineland
- Five Feet Apart, a 2019 romantic drama film starring Cole Sprouse and Haley Lu Richardson
- Orla Tinsley: Warrior, a 2018 documentary film about CF campaigner Orla Tinsley
- The performance art of Martin O’Brien
- Continent Chasers, a traveller and CF patient documenting travel and CF blogs, continentchasers.com
Notes[edit]
- ^ The Cystic Fibrosis Foundation recommends a diagnosis of cystic fibrosis for anyone suspected of cystic fibrosis (positive newborn screen, symptoms of cystic fibrosis, or a family history of cystic fibrosis) with sweat chloride above 60 millimoles/liter. Those with less than 30 millimoles/liter sweat chloride are unlikely to develop cystic fibrosis. For people with intermediate sweat chloride between 30 and 59 millimoles/liter, they recommend additional genetic testing.[68]
References[edit]
- ^ a b c d e f g h i j k l m n o p q r s t u O’Sullivan BP, Freedman SD (May 2009). «Cystic fibrosis». Lancet. 373 (9678): 1891–904. doi:10.1016/s0140-6736(09)60327-5. PMID 19403164. S2CID 46011502.
- ^ Allen JL, Panitch HB, Rubenstein RC (2016). Cystic Fibrosis. CRC Press. p. 92. ISBN 9781439801826. Archived from the original on 8 September 2017.
- ^ a b c d Massie J, Delatycki MB (December 2013). «Cystic fibrosis carrier screening». Paediatric Respiratory Reviews. 14 (4): 270–5. doi:10.1016/j.prrv.2012.12.002. PMID 23466339.
- ^ a b Ong T, Ramsey BW (September 2015). «Update in Cystic Fibrosis 2014». American Journal of Respiratory and Critical Care Medicine. 192 (6): 669–75. doi:10.1164/rccm.201504-0656UP. PMID 26371812.
- ^ Sencen L. «Cystic Fibrosis». NORD (National Organization for Rare Disorders). Retrieved 29 July 2022.
- ^ «Orphanet: Cystic fibrosis». www.orpha.net. Retrieved 29 July 2022.
- ^ a b c Hodson M, Geddes D, Bush A, eds. (2012). Cystic Fibrosis (3rd ed.). London: Hodder Arnold. p. 3. ISBN 978-1-4441-1369-3. Archived from the original on 8 September 2017.
- ^ Buckingham L (2012). Molecular Diagnostics: Fundamentals, Methods and Clinical Applications (2nd ed.). Philadelphia: F.A. Davis Co. p. 351. ISBN 978-0-8036-2975-2. Archived from the original on 8 September 2017.
- ^ Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D (January 2004). «Cystic fibrosis adult care: consensus conference report». Chest. 125 (1 Suppl): 1S–39S. CiteSeerX 10.1.1.562.1904. doi:10.1378/chest.125.1_suppl.1S. PMID 14734689.
- ^ a b Warnock L, Gates A (December 2015). «Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis». The Cochrane Database of Systematic Reviews. 2015 (12): CD001401. doi:10.1002/14651858.CD001401.pub3. PMC 6768986. PMID 26688006.
- ^ Nazareth D, Walshaw M (October 2013). «Coming of age in cystic fibrosis — transition from paediatric to adult care». Clinical Medicine. 13 (5): 482–6. doi:10.7861/clinmedicine.13-5-482. PMC 4953800. PMID 24115706.
- ^ a b Andersen DH (1938). «Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study». Am. J. Dis. Child. 56 (2): 344–99. doi:10.1001/archpedi.1938.01980140114013.
- ^ a b c d Egan, Schechter & Voynow 2020, «Clinical Manifestations».
- ^ a b c d e f Egan, Schechter & Voynow 2020, «Respiratory Tract».
- ^ Shteinberg M, Haq IJ, Polineni D, Davies JC (June 2021). «Cystic fibrosis». Lancet. 397 (10290): 2195–2211. doi:10.1016/S0140-6736(20)32542-3. PMID 34090606. S2CID 235327978.
- ^ Reaves J, Wallace G (2010). «Unexplained bruising: weighing the pros and cons of possible causes». Consultant for Pediatricians. 9: 201–2. Archived from the original on 22 February 2020. Retrieved 22 February 2020.
- ^ Flume PA, Mogayzel PJ, Robinson KA, Rosenblatt RL, Quittell L, Marshall BC (August 2010). «Cystic fibrosis pulmonary guidelines: pulmonary complications: hemoptysis and pneumothorax». American Journal of Respiratory and Critical Care Medicine. 182 (3): 298–306. doi:10.1164/rccm.201002-0157OC. PMID 20675678.
- ^ a b c d e f g h i j k l m n o p q r s t Mitchell RS, Kumar V, Robbins SL, et al. (2007). Robbins Basic Pathology. Saunders/Elsevier. p. 1253, 1254. ISBN 978-1-4160-2973-1.
- ^ a b c d Rowe SM, Miller S, Sorscher EJ (May 2005). «Cystic fibrosis». The New England Journal of Medicine. 352 (19): 1992–2001. doi:10.1056/NEJMra043184. PMID 15888700.
- ^ Saiman L, Siegel J (January 2004). «Infection control in cystic fibrosis». Clinical Microbiology Reviews. 17 (1): 57–71. doi:10.1128/CMR.17.1.57-71.2004. PMC 321464. PMID 14726455.
- ^ Girón RM, Domingo D, Buendía B, Antón E, Ruiz-Velasco LM, Ancochea J (October 2005). «[Nontuberculous mycobacteria in patients with cystic fibrosis]». Archivos de Bronconeumologia (in Spanish). 41 (10): 560–5. doi:10.1016/S1579-2129(06)60283-8. PMID 16266669.
- ^ Franco LP, Camargos PA, Becker HM, Guimarães RE (2009). «Nasal endoscopic evaluation of children and adolescents with cystic fibrosis». Brazilian Journal of Otorhinolaryngology. 75 (6): 806–13. doi:10.1590/S1808-86942009000600006. PMC 9446041. PMID 20209279.
- ^ Maldonado M, Martínez A, Alobid I, Mullol J (December 2004). «The antrochoanal polyp». Rhinology. 42 (4): 178–82. PMID 15626248.
- ^ Ramsey B, Richardson MA (September 1992). «Impact of sinusitis in cystic fibrosis». The Journal of Allergy and Clinical Immunology. 90 (3 Pt 2): 547–52. doi:10.1016/0091-6749(92)90183-3. PMID 1527348.
- ^ Kulczycki LL, Shwachman H (August 1958). «Studies in cystic fibrosis of the pancreas; occurrence of rectal prolapse». The New England Journal of Medicine. 259 (9): 409–12. doi:10.1056/NEJM195808282590901. PMID 13578072.
- ^ Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS (September 1998). «Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis». The New England Journal of Medicine. 339 (10): 653–8. doi:10.1056/NEJM199809033391002. PMID 9725922.
- ^ a b c Assis DN, Freedman SD (March 2016). «Gastrointestinal Disorders in Cystic Fibrosis». Clinics in Chest Medicine (Review). 37 (1): 109–118. doi:10.1016/j.ccm.2015.11.004. PMID 26857772.
- ^ Malfroot A, Dab I (November 1991). «New insights on gastro-oesophageal reflux in cystic fibrosis by longitudinal follow up». Archives of Disease in Childhood. 66 (11): 1339–1345. doi:10.1136/adc.66.11.1339. PMC 1793275. PMID 1755649.
- ^ a b c d Carroll W, Green J, Gilchrist FJ (December 2021). «Interventions for preventing distal intestinal obstruction syndrome (DIOS) in cystic fibrosis». The Cochrane Database of Systematic Reviews. 2021 (12): CD012619. doi:10.1002/14651858.CD012619.pub3. PMC 8693853. PMID 34936085.
- ^ Williams SG, Westaby D, Tanner MS, Mowat AP (October 1992). «Liver and biliary problems in cystic fibrosis». British Medical Bulletin. 48 (4): 877–92. doi:10.1093/oxfordjournals.bmb.a072583. PMID 1458306.
- ^ Colombo C, Russo MC, Zazzeron L, Romano G (July 2006). «Liver disease in cystic fibrosis». Journal of Pediatric Gastroenterology and Nutrition. 43 (Suppl 1): S49-55. doi:10.1097/01.mpg.0000226390.02355.52. PMID 16819402. S2CID 27836468.
- ^ Egan, Schechter & Voynow 2020, «Biliary Tract».
- ^ Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS, Seaquist ER (August 1994). «Insulin sensitivity in cystic fibrosis». Diabetes. 43 (8): 1020–6. doi:10.2337/diabetes.43.8.1020. PMID 8039595.
- ^ a b c de Aragão Dantas Alves C, Aguiar RA, Alves AC, Santana MA (2007). «Diabetes mellitus in patients with cystic fibrosis». Jornal Brasileiro de Pneumologia. 33 (2): 213–21. doi:10.1590/S1806-37132007000200017. PMID 17724542.
- ^ Haworth CS, Selby PL, Webb AK, Dodd ME, Musson H, McL Niven R, et al. (November 1999). «Low bone mineral density in adults with cystic fibrosis». Thorax. 54 (11): 961–7. doi:10.1136/thx.54.11.961. PMC 1745400. PMID 10525552.
- ^ McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD (October 2000). «Fertility in men with cystic fibrosis: an update on current surgical practices and outcomes». Chest. 118 (4): 1059–62. doi:10.1378/chest.118.4.1059. PMID 11035677.
- ^ Chen H, Ruan YC, Xu WM, Chen J, Chan HC (2012). «Regulation of male fertility by CFTR and implications in male infertility». Human Reproduction Update. 18 (6): 703–13. doi:10.1093/humupd/dms027. PMID 22709980.
- ^ Augarten A, Yahav Y, Kerem BS, Halle D, Laufer J, Szeinberg A, et al. (November 1994). «Congenital bilateral absence of vas deferens in the absence of cystic fibrosis». Lancet. 344 (8935): 1473–4. doi:10.1016/S0140-6736(94)90292-5. PMID 7968122. S2CID 28860665.
- ^ Gilljam M, Antoniou M, Shin J, Dupuis A, Corey M, Tullis DE (July 2000). «Pregnancy in cystic fibrosis. Fetal and maternal outcome». Chest. 118 (1): 85–91. doi:10.1378/chest.118.1.85. PMID 10893364. S2CID 32289370.
- ^ Guimbellot J, Sharma J, Rowe SM (November 2017). «Toward inclusive therapy with CFTR modulators: Progress and challenges». Pediatric Pulmonology. 52 (S48): S4–S14. doi:10.1002/ppul.23773. PMC 6208153. PMID 28881097.
- ^ Sharma J, Keeling KM, Rowe SM (August 2020). «Pharmacological approaches for targeting cystic fibrosis nonsense mutations». European Journal of Medicinal Chemistry. 200: 112436. doi:10.1016/j.ejmech.2020.112436. PMC 7384597. PMID 32512483.
- ^ Bobadilla JL, Macek M, Fine JP, Farrell PM (June 2002). «Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening». Human Mutation. 19 (6): 575–606. doi:10.1002/humu.10041. PMID 12007216. S2CID 35428054.
- ^ a b c Elborn JS (November 2016). «Cystic fibrosis». Lancet. 388 (10059): 2519–2531. doi:10.1016/S0140-6736(16)00576-6. PMID 27140670. S2CID 20948144.
- ^ Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, et al. (July 1998). «An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton». The Journal of Biological Chemistry. 273 (31): 19797–801. doi:10.1074/jbc.273.31.19797. PMID 9677412.
- ^ Travaglini KJ, Krasnow MA (August 2018). «Profile of an unknown airway cell». Nature. 560 (7718): 313–314. Bibcode:2018Natur.560..313T. doi:10.1038/d41586-018-05813-7. PMID 30097657.
- ^ a b Edwards QT, Seibert D, Macri C, Covington C, Tilghman J (November 2004). «Assessing ethnicity in preconception counseling: genetics—what nurse practitioners need to know». Journal of the American Academy of Nurse Practitioners. 16 (11): 472–80. doi:10.1111/j.1745-7599.2004.tb00426.x. PMID 15617360. S2CID 7644129.
- ^ Wang XR, Li C (May 2014). «Decoding F508del misfolding in cystic fibrosis». Biomolecules. 4 (2): 498–509. doi:10.3390/biom4020498. PMC 4101494. PMID 24970227.
- ^ Childers M, Eckel G, Himmel A, Caldwell J (2007). «A new model of cystic fibrosis pathology: lack of transport of glutathione and its thiocyanate conjugates». Medical Hypotheses. 68 (1): 101–12. doi:10.1016/j.mehy.2006.06.020. PMID 16934416.
- ^ Xu Y, Szép S, Lu Z (December 2009). «The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases». Proceedings of the National Academy of Sciences of the United States of America. 106 (48): 20515–9. Bibcode:2009PNAS..10620515X. doi:10.1073/pnas.0911412106. PMC 2777967. PMID 19918082.
- ^ Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Nauseef WM, et al. (January 2007). «A novel host defense system of airways is defective in cystic fibrosis». American Journal of Respiratory and Critical Care Medicine. 175 (2): 174–83. doi:10.1164/rccm.200607-1029OC. PMC 2720149. PMID 17082494.
- ^ Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M (January 2007). «The lactoperoxidase system links anion transport to host defense in cystic fibrosis». FEBS Letters. 581 (2): 271–8. doi:10.1016/j.febslet.2006.12.025. PMC 1851694. PMID 17204267.
- ^ Haq IJ, Gray MA, Garnett JP, Ward C, Brodlie M (March 2016). «Airway surface liquid homeostasis in cystic fibrosis: pathophysiology and therapeutic targets». Thorax. 71 (3): 284–287. doi:10.1136/thoraxjnl-2015-207588. PMID 26719229.
- ^ Verkman AS, Song Y, Thiagarajah JR (January 2003). «Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease». American Journal of Physiology. Cell Physiology. 284 (1): C2-15. doi:10.1152/ajpcell.00417.2002. PMID 12475759. S2CID 11790119.
- ^ Marieb EN, Hoehn K, Hutchinson M (2014). «22: The Respiratory System». Human Anatomy and Physiology. Pearson Education. p. 906. ISBN 978-0805361179.
- ^ a b Saiman L (2004). «Microbiology of early CF lung disease». Paediatric Respiratory Reviews. 5 (Suppl A): S367-9. doi:10.1016/S1526-0542(04)90065-6. PMID 14980298.
- ^ a b Khanolkar RA, Clark ST, Wang PW, et al. (2020). «Ecological Succession of Polymicrobial Communities in the Cystic Fibrosis Airways». mSystems. 5 (6): e00809-20. doi:10.1128/mSystems.00809-20. PMC 7716390. PMID 33262240.
- ^ a b Lorè NI, Cigana C, Riva C, De Fino I, Nonis A, Spagnuolo L, et al. (May 2016). «IL-17A impairs host tolerance during airway chronic infection by Pseudomonas aeruginosa». Scientific Reports. 6: 25937. Bibcode:2016NatSR…625937L. doi:10.1038/srep25937. PMC 4870500. PMID 27189736.
- ^ Tümmler B, Koopmann U, Grothues D, Weissbrodt H, Steinkamp G, von der Hardt H (June 1991). «Nosocomial acquisition of Pseudomonas aeruginosa by cystic fibrosis patients». Journal of Clinical Microbiology. 29 (6): 1265–7. Bibcode:1991JPoSA..29.1265A. doi:10.1002/pola.1991.080290905. PMC 271975. PMID 1907611.
- ^ Centers for Disease Control Prevention (CDC) (June 1993). «Pseudomonas cepacia at summer camps for persons with cystic fibrosis». MMWR. Morbidity and Mortality Weekly Report. 42 (23): 456–9. PMID 7684813.
- ^ Pegues DA, Carson LA, Tablan OC, FitzSimmons SC, Roman SB, Miller JM, Jarvis WR (May 1994). «Acquisition of Pseudomonas cepacia at summer camps for patients with cystic fibrosis. Summer Camp Study Group». The Journal of Pediatrics. 124 (5 Pt 1): 694–702. doi:10.1016/S0022-3476(05)81357-5. PMID 7513755.
- ^ Pankhurst CL, Philpott-Howard J (April 1996). «The environmental risk factors associated with medical and dental equipment in the transmission of Burkholderia (Pseudomonas) cepacia in cystic fibrosis patients». The Journal of Hospital Infection. 32 (4): 249–55. doi:10.1016/S0195-6701(96)90035-3. PMID 8744509.
- ^ Jones AM, Govan JR, Doherty CJ, Dodd ME, Isalska BJ, Stanbridge TN, Webb AK (June 2003). «Identification of airborne dissemination of epidemic multiresistant strains of Pseudomonas aeruginosa at a CF centre during a cross infection outbreak». Thorax. 58 (6): 525–7. doi:10.1136/thorax.58.6.525. PMC 1746694. PMID 12775867.
- ^ Høiby N (June 1995). «Isolation and treatment of cystic fibrosis patients with lung infections caused by Pseudomonas (Burkholderia) cepacia and multiresistant Pseudomonas aeruginosa». The Netherlands Journal of Medicine. 46 (6): 280–7. doi:10.1016/0300-2977(95)00020-N. PMID 7643943.
- ^ a b Pihet M, Carrere J, Cimon B, Chabasse D, Delhaes L, Symoens F, Bouchara JP (June 2009). «Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis—a review». Medical Mycology. 47 (4): 387–97. doi:10.1080/13693780802609604. PMID 19107638.
- ^ Rapaka RR, Kolls JK (2009). «Pathogenesis of allergic bronchopulmonary aspergillosis in cystic fibrosis: current understanding and future directions». Medical Mycology. 47 (Suppl 1): S331-7. doi:10.1080/13693780802266777. PMID 18668399.
- ^ «Newborn Screening for CF». Cystic Fibrosis Foundation. Retrieved 25 January 2022.
- ^ a b Egan, Schechter & Voynow 2020, «Diagnosis and Assessment».
- ^ Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, Howenstine M, McColley SA, Rock M, Rosenfeld M, Sermet-Gaudelus I, Southern KW, Marshall BC, Sosnay PR (February 2017). «Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation». J Pediatr. 181S: S4–S15.e1. doi:10.1016/j.jpeds.2016.09.064. PMID 28129811. S2CID 206410545.
- ^ Minarowski Ł, Sands D, Minarowska A, Karwowska A, Sulewska A, Gacko M, Chyczewska E (2008). «Thiocyanate concentration in saliva of cystic fibrosis patients». Folia Histochemica et Cytobiologica. 46 (2): 245–6. doi:10.2478/v10042-008-0037-0. PMID 18519245.
- ^ Stern RC (February 1997). «The diagnosis of cystic fibrosis». The New England Journal of Medicine. 336 (7): 487–91. doi:10.1056/NEJM199702133360707. PMID 9017943.
- ^ Ross LF (September 2008). «Newborn screening for cystic fibrosis: a lesson in public health disparities». The Journal of Pediatrics. 153 (3): 308–13. doi:10.1016/j.jpeds.2008.04.061. PMC 2569148. PMID 18718257.
- ^ Assael BM, Castellani C, Ocampo MB, Iansa P, Callegaro A, Valsecchi MG (September 2002). «Epidemiology and survival analysis of cystic fibrosis in an area of intense neonatal screening over 30 years». American Journal of Epidemiology. 156 (5): 397–401. doi:10.1093/aje/kwf064. PMID 12196308.
- ^ Hoch H, Sontag MK, Scarbro S, Juarez-Colunga E, McLean C, Kempe A, Sagel SD (November 2018). «Clinical outcomes in U.S. infants with cystic fibrosis from 2001 to 2012». Pediatric Pulmonology. 53 (11): 1492–1497. doi:10.1002/ppul.24165. PMID 30259702. S2CID 52845580.
- ^ Barben, Jürg, et al. (2017). «The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe». Journal of Cystic Fibrosis. 16 (2): 207–13. doi:10.1016/j.jcf.2016.12.012. PMID 28043799.
- ^ «Carrier Screening in the Age of Genomic Medicine». American College of Obstetricians and Gynecologists. 2017. Archived from the original on 25 February 2017. Retrieved 22 February 2020.
- ^ Elias S, Annas GJ, Simpson JL (April 1991). «Carrier screening for cystic fibrosis: implications for obstetric and gynecologic practice». American Journal of Obstetrics and Gynecology. 164 (4): 1077–83. doi:10.1016/0002-9378(91)90589-j. PMID 2014829.
- ^ Tabor A, Philip J, Madsen M, Bang J, Obel EB, Nørgaard-Pedersen B (June 1986). «Randomised controlled trial of genetic amniocentesis in 4606 low-risk women». Lancet. 1 (8493): 1287–93. doi:10.1016/S0140-6736(86)91218-3. PMID 2423826. S2CID 31237495.
- ^ Eddleman KA, Malone FD, Sullivan L, Dukes K, Berkowitz RL, Kharbutli Y, et al. (November 2006). «Pregnancy loss rates after midtrimester amniocentesis». Obstetrics and Gynecology. 108 (5): 1067–72. doi:10.1097/01.AOG.0000240135.13594.07. PMID 17077226. S2CID 19081825.
- ^ Davis LB, Champion SJ, Fair SO, Baker VL, Garber AM (April 2010). «A cost-benefit analysis of preimplantation genetic diagnosis for carrier couples of cystic fibrosis». Fertility and Sterility. 93 (6): 1793–804. doi:10.1016/j.fertnstert.2008.12.053. PMID 19439290.
- ^ a b «Abstracts from the 25th Italian Congress of Cystic Fibrosis and the 15th National Congress of Cystic Fibrosis Italian Society : Assago, Milan. 10 — 12 October 2019». Italian Journal of Pediatrics. 46 (Suppl 1): 32. April 2020. doi:10.1186/s13052-020-0790-z. PMC 7110616. PMID 32234058.
- ^
- ^ Davies JC, Alton EW, Bush A (December 2007). «Cystic fibrosis». BMJ. 335 (7632): 1255–9. doi:10.1136/bmj.39391.713229.AD. PMC 2137053. PMID 18079549.
- ^ Hayes D, Wilson KC, Krivchenia K, Hawkins SM, Balfour-Lynn IM, Gozal D, et al. (February 2019). «Home Oxygen Therapy for Children. An Official American Thoracic Society Clinical Practice Guideline». American Journal of Respiratory and Critical Care Medicine. 199 (3): e5–e23. doi:10.1164/rccm.201812-2276ST. PMC 6802853. PMID 30707039.
- ^ Coffey MJ, Garg M, Homaira N, Jaffe A, Ooi CY (January 2020). «Probiotics for people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 1 (1): CD012949. doi:10.1002/14651858.CD012949.pub2. PMC 6984633. PMID 31962375.
- ^ Pai VB, Nahata MC (October 2001). «Efficacy and safety of aerosolized tobramycin in cystic fibrosis». Pediatric Pulmonology. 32 (4): 314–27. doi:10.1002/ppul.1125. PMID 11568993. S2CID 30108514.
- ^ Westerman EM, Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG (March 2004). «Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study». Journal of Cystic Fibrosis. 3 (1): 23–8. doi:10.1016/j.jcf.2003.12.005. PMID 15463883.
- ^ McCoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch-Bogart GZ, Montgomery AB (November 2008). «Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis». American Journal of Respiratory and Critical Care Medicine. 178 (9): 921–8. doi:10.1164/rccm.200712-1804OC. PMC 2577727. PMID 18658109.
- ^ Ryan G, Singh M, Dwan K (March 2011). «Inhaled antibiotics for long-term therapy in cystic fibrosis». The Cochrane Database of Systematic Reviews (3): CD001021. doi:10.1002/14651858.CD001021.pub2. PMID 21412868.
- ^ «Quinsair (levofloxacin)». European Medicines Agency. Archived from the original on 26 December 2016. Retrieved 26 December 2016.
- ^ Langton Hewer SC, Smyth AR (April 2017). «Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis» (PDF). The Cochrane Database of Systematic Reviews. 4 (4): CD004197. doi:10.1002/14651858.CD004197.pub5. PMC 6478104. PMID 28440853. Archived from the original (PDF) on 24 February 2019. Retrieved 22 January 2019.
- ^ Smith S, Ratjen F, Remmington T, Waters V (May 2020). «Combination antimicrobial susceptibility testing for acute exacerbations in chronic infection of Pseudomonas aeruginosa in cystic fibrosis». The Cochrane Database of Systematic Reviews. 5 (4): CD006961. doi:10.1002/14651858.CD006961.pub5. PMC 7387858. PMID 32412092.
- ^ Hansen CR, Pressler T, Koch C, Høiby N (March 2005). «Long-term azitromycin treatment of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection; an observational cohort study». Journal of Cystic Fibrosis. 4 (1): 35–40. doi:10.1016/j.jcf.2004.09.001. PMID 15752679.
- ^ Tan KH, Mulheran M, Knox AJ, Smyth AR (March 2003). «Aminoglycoside prescribing and surveillance in cystic fibrosis». American Journal of Respiratory and Critical Care Medicine. 167 (6): 819–23. doi:10.1164/rccm.200109-012CC. PMID 12623858.
- ^ «Antibiotic resistance». Fact Sheet. World Health Organization. 31 July 2020. Retrieved 24 June 2022.
- ^ Hurley MN, Smith S, Forrester DL, Smyth AR (July 2020). «Antibiotic adjuvant therapy for pulmonary infection in cystic fibrosis». The Cochrane Database of Systematic Reviews. 7 (9): CD008037. doi:10.1002/14651858.CD008037.pub4. PMC 8407502. PMID 32671834.
- ^ Lord R, Jones AM, Horsley A (April 2020). «Antibiotic treatment for Burkholderia cepacia complex in people with cystic fibrosis experiencing a pulmonary exacerbation». The Cochrane Database of Systematic Reviews. 2020 (4): CD009529. doi:10.1002/14651858.CD009529.pub4. PMC 7117566. PMID 32239690.
- ^ Waters V, Ratjen F (June 2020). «Antibiotic treatment for nontuberculous mycobacteria lung infection in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 6 (6): CD010004. doi:10.1002/14651858.CD010004.pub5. PMC 7389742. PMID 32521055.
- ^ Kuver R, Lee SP (April 2006). «Hypertonic saline for cystic fibrosis». The New England Journal of Medicine. 354 (17): 1848–51, author reply 1848–51. doi:10.1056/NEJMc060351. PMID 16642591.
- ^ Lieberman J (July 1968). «Dornase aerosol effect on sputum viscosity in cases of cystic fibrosis». JAMA. 205 (5): 312–3. doi:10.1001/jama.205.5.312. PMID 5694947.
- ^ a b Yang C, Montgomery M, et al. (Cochrane Cystic Fibrosis and Genetic Disorders Group) (March 2021). «Dornase alfa for cystic fibrosis». The Cochrane Database of Systematic Reviews. 2021 (3): CD001127. doi:10.1002/14651858.CD001127.pub5. PMC 8094421. PMID 33735508.
- ^ Kellerman D, Rossi Mospan A, Engels J, Schaberg A, Gorden J, Smiley L (August 2008). «Denufosol: a review of studies with inhaled P2Y(2) agonists that led to Phase 3». Pulmonary Pharmacology & Therapeutics. 21 (4): 600–7. doi:10.1016/j.pupt.2007.12.003. PMID 18276176.
- ^ a b Balfour-Lynn IM, Welch K, Smith S (July 2019). «Inhaled corticosteroids for cystic fibrosis». The Cochrane Database of Systematic Reviews. 7 (4): CD001915. doi:10.1002/14651858.CD001915.pub6. PMC 6609325. PMID 31271656.
- ^ Burgess L, Southern KW (August 2014). Burgess L (ed.). «Pneumococcal vaccines for cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (8): CD008865. doi:10.1002/14651858.CD008865.pub3. PMID 25093421.
- ^ Dharmaraj P, Smyth RL (March 2014). «Vaccines for preventing influenza in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 2021 (3): CD001753. doi:10.1002/14651858.CD001753.pub3. PMC 7066935. PMID 24604671.
- ^ a b c Whiting P, Al M, Burgers L, Westwood M, Ryder S, Hoogendoorn M, et al. (March 2014). «Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis». Health Technology Assessment. 18 (18): 1–106. doi:10.3310/hta18180. PMC 4780965. PMID 24656117.
- ^ Wainwright CE (October 2014). «Ivacaftor for patients with cystic fibrosis». Expert Review of Respiratory Medicine. 8 (5): 533–8. doi:10.1586/17476348.2014.951333. PMID 25148205. S2CID 39537446.
- ^ Office of the Commissioner. «Press Announcements — FDA approves new treatment for cystic fibrosis». www.fda.gov. Archived from the original on 18 January 2017. Retrieved 16 January 2017.
- ^ «FDA approves another Vertex drug for treatment of cystic fibrosis — The Boston Globe». The Boston Globe.
- ^ «Tezacaftor (VX-661) for Cystic Fibrosis — Cystic Fibrosis News Today». Archived from the original on 29 September 2018. Retrieved 23 December 2018.
- ^ a b «Trikafta (elexacaftor, ivacaftor and tezacaftor) FDA Approval History». Drugs.com.
- ^ Office of the Commissioner (24 March 2020). «FDA approves new breakthrough therapy for cystic fibrosis». FDA. Retrieved 28 April 2022.
- ^ «FDA Accepts Vertex Application for Expansion of Trikafta to Include Children ages 6-11 | Cystic Fibrosis Foundation». www.cff.org. Retrieved 28 April 2022.
- ^ «NHS England » Landmark NHS deal to open up access to life-changing cystic fibrosis drug». www.england.nhs.uk. Retrieved 8 August 2021.
- ^ Office of the Commissioner (24 March 2020). «FDA approves new breakthrough therapy for cystic fibrosis». FDA. Retrieved 12 August 2020.
- ^ a b «Cystic Fibrosis Foundation statement on FDA approval of Trikafta™, the first triple-combination therapy for the most common CF mutation». www.cff.org. Bethesda, Md.: Cystic Fibrosis Foundation. Retrieved 12 August 2020.
- ^ a b Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. (November 2019). «Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele». The New England Journal of Medicine. 381 (19): 1809–1819. doi:10.1056/NEJMoa1908639. PMC 7282384. PMID 31697873.
- ^ Ridley K, Condren M (1 April 2020). «Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy». The Journal of Pediatric Pharmacology and Therapeutics. 25 (3): 192–197. doi:10.5863/1551-6776-25.3.192. PMC 7134581. PMID 32265602.
- ^ «Vertex prices cystic fibrosis combo treatment at $311,000-per-year». Reuters. 21 October 2019. Retrieved 23 October 2019.
- ^ Good News Network (3 November 2019). «FDA Approves the First New Cystic Fibrosis Treatment in Decades». Good News Network. Retrieved 12 August 2020.
- ^ Cheng K, Ashby D, Smyth RL (September 2017). «Ursodeoxycholic acid for cystic fibrosis-related liver disease». The Cochrane Database of Systematic Reviews. 9 (4): CD000222. doi:10.1002/14651858.CD000222.pub4. PMC 6483662. PMID 28891588.
- ^ de Vries JJ, Chang AB, Bonifant CM, Shevill E, Marchant JM (August 2018). «Vitamin A and beta (β)-carotene supplementation for cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (8): CD006751. doi:10.1002/14651858.CD006751.pub5. PMC 6513379. PMID 30091146.
- ^ Ferguson JH, Chang AB (May 2014). «Vitamin D supplementation for cystic fibrosis». The Cochrane Database of Systematic Reviews (5): CD007298. doi:10.1002/14651858.CD007298.pub4. PMID 24823922.
- ^ Okebukola PO, Kansra S, Barrett J (September 2020). «Vitamin E supplementation in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 2020 (9): CD009422. doi:10.1002/14651858.CD009422.pub4. PMC 8406985. PMID 32892350.
- ^ Jagannath VA, Thaker V, Chang AB, Price AI (June 2020). «Vitamin K supplementation for cystic fibrosis». The Cochrane Database of Systematic Reviews. John Wiley & sons, Ltd. 6 (6): CD008482. doi:10.1002/14651858.CD008482.pub6. PMC 7272115. PMID 32497260.
- ^ Watson H, Stackhouse C (April 2020). «Omega-3 fatty acid supplementation for cystic fibrosis». The Cochrane Database of Systematic Reviews. 4 (4): CD002201. doi:10.1002/14651858.CD002201.pub6. PMC 7147930. PMID 32275788.
- ^ McIlwaine M, Button B, Nevitt SJ (November 2019). «Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 2019 (11). doi:10.1002/14651858.CD003147.pub5. PMC 6953327. PMID 31774149.
- ^ Andersen JB, Qvist J, Kann T (October 1979). «Recruiting collapsed lung through collateral channels with positive end-expiratory pressure». Scandinavian Journal of Respiratory Diseases. 60 (5): 260–6. PMID 392747.
- ^ Groth S, Stafanger G, Dirksen H, Andersen JB, Falk M, Kelstrup M (July 1985). «Positive expiratory pressure (PEP-mask) physiotherapy improves ventilation and reduces volume of trapped gas in cystic fibrosis». Bulletin Européen de Physiopathologie Respiratoire. 21 (4): 339–43. PMID 3899222.
- ^ a b c Moran F, Bradley JM, Piper AJ (February 2017). «Non-invasive ventilation for cystic fibrosis». The Cochrane Database of Systematic Reviews. 2 (2): CD002769. doi:10.1002/14651858.CD002769.pub5. PMC 6464053. PMID 28218802.
- ^ Moran F, Bradley JM, Piper AJ (February 2017). «Non-invasive ventilation for cystic fibrosis». The Cochrane Database of Systematic Reviews. 2 (2): CD002769. doi:10.1002/14651858.CD002769.pub5. PMC 6464053. PMID 28218802.
- ^ «Tracheostomy Why it’s used». NHS. 3 October 2018. Retrieved 10 May 2020.
- ^ Molnar H. «Types of Tracheostomy Tubes».
- ^ Huth MM, Zink KA, Van Horn NR (2005). «The effects of massage therapy in improving outcomes for youth with cystic fibrosis: an evidence review». Pediatric Nursing. 31 (4): 328–32. PMID 16229132.
- ^ Leinwand MJ (28 December 2019). Windle ML, Odim J (eds.). «Surgical Treatment of Infections of the Lung, Pleura, and Mediastinum». Medscape. Archived from the original on 5 October 2016.
- ^ Amin R, Noone PG, Ratjen F (December 2012). «Chemical pleurodesis versus surgical intervention for persistent and recurrent pneumothoraces in cystic fibrosis». The Cochrane Database of Systematic Reviews. 12 (12): CD007481. doi:10.1002/14651858.CD007481.pub3. PMC 7208277. PMID 23235645.
- ^ Fridell JA, Vianna R, Kwo PY, Howenstine M, Sannuti A, Molleston JP, et al. (October 2005). «Simultaneous liver and pancreas transplantation in patients with cystic fibrosis». Transplantation Proceedings. 37 (8): 3567–9. doi:10.1016/j.transproceed.2005.09.091. PMID 16298663.
- ^ Belkin RA, Henig NR, Singer LG, Chaparro C, Rubenstein RC, Xie SX, et al. (March 2006). «Risk factors for death of patients with cystic fibrosis awaiting lung transplantation». American Journal of Respiratory and Critical Care Medicine. 173 (6): 659–66. doi:10.1164/rccm.200410-1369OC. PMC 2662949. PMID 16387803.
- ^ a b «Cystic Fibrosis — Pediatrics». Merck Manuals Professional Edition. Retrieved 12 August 2020.
- ^ Somaraju UR, Solis-Moya A (August 2020). «Pancreatic enzyme replacement therapy for people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (9): CD008227. doi:10.1002/14651858.CD008227.pub4. PMC 8094413. PMID 32761612.
- ^ Skolnik K, Levy RD, Wilcox PG, Quon BS (November 2016). «Coronary artery disease in cystic fibrosis: An emerging concern?». Journal of Cystic Fibrosis. 15 (6): e70–e71. doi:10.1016/j.jcf.2016.09.010. PMID 27751792.
- ^ Zirbes J, Milla CE (September 2009). «Cystic fibrosis related diabetes». Paediatric Respiratory Reviews. 10 (3): 118–23, quiz 123. doi:10.1016/j.prrv.2009.04.004. PMID 19651382.
- ^ Onady GM, Stolfi A (October 2020). «Drug treatments for managing cystic fibrosis-related diabetes». The Cochrane Database of Systematic Reviews. 2020 (10): CD004730. doi:10.1002/14651858.CD004730.pub5. PMC 8094754. PMID 33075159.
- ^ Amin R, Jahnke N, Waters V (March 2020). «Antibiotic treatment for Stenotrophomonas maltophilia in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 3 (4): CD009249. doi:10.1002/14651858.CD009249.pub5. PMC 7080526. PMID 32189337.
- ^ a b c Conwell LS, Chang AB (March 2014). «Bisphosphonates for osteoporosis in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 2014 (3): CD002010. doi:10.1002/14651858.CD002010.pub4. PMC 6718208. PMID 24627308.
- ^ Hardin DS, Rice J, Ahn C, Ferkol T, Howenstine M, Spears S, et al. (March 2005). «Growth hormone treatment enhances nutrition and growth in children with cystic fibrosis receiving enteral nutrition». The Journal of Pediatrics. 146 (3): 324–8. doi:10.1016/j.jpeds.2004.10.037. PMID 15756212.
- ^ Marks SC, Kissner DG (1997). «Management of sinusitis in adult cystic fibrosis». American Journal of Rhinology. 11 (1): 11–4. doi:10.2500/105065897781446810. PMID 9065342. S2CID 5606258.
- ^ Phillipson GT, Petrucco OM, Matthews CD (February 2000). «Congenital bilateral absence of the vas deferens, cystic fibrosis mutation analysis and intracytoplasmic sperm injection». Human Reproduction. 15 (2): 431–5. doi:10.1093/humrep/15.2.431. PMID 10655317.
- ^ Ciofu O, Lykkesfeldt J (August 2014). «Antioxidant supplementation for lung disease in cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (8): CD007020. doi:10.1002/14651858.CD007020.pub3. PMC 6777741. PMID 25102015.
- ^ a b Radtke, Thomas; Smith, Sherie; Nevitt, Sarah J.; Hebestreit, Helge; Kriemler, Susi (9 August 2022). «Physical activity and exercise training in cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (8): CD002768. doi:10.1002/14651858.CD002768.pub5. ISSN 1469-493X. PMC 9361297. PMID 35943025.
- ^ Ganesan P, Schmiedge J, Manchaiah V, Swapna S, Dhandayutham S, Kothandaraman PP (April 2018). «Ototoxicity: A Challenge in Diagnosis and Treatment». Journal of Audiology & Otology. 22 (2): 59–68. doi:10.7874/jao.2017.00360. PMC 5894487. PMID 29471610.
- ^ Green J, Gilchrist FJ, Carroll W (June 2018). «Interventions for preventing distal intestinal obstruction syndrome (DIOS) in cystic fibrosis». The Cochrane Database of Systematic Reviews. 6 (6): CD012619. doi:10.1002/14651858.cd012619. PMC 6478257. PMID 29894558.
- ^ Davis PB (March 2006). «Cystic fibrosis since 1938». American Journal of Respiratory and Critical Care Medicine. 173 (5): 475–82. doi:10.1164/rccm.200505-840OE. PMID 16126935. S2CID 1770759.
- ^ MacKenzie T, Gifford AH, Sabadosa KA, Quinton HB, Knapp EA, Goss CH, Marshall BC (August 2014). «Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation patient registry». Annals of Internal Medicine. 161 (4): 233–41. doi:10.7326/m13-0636. PMC 4687404. PMID 25133359.
- ^ «Canadian Cystic Fibrosis Patient Data Registry Report» (PDF). Canadian Cystic Fibrosis Foundation. 2007. Archived from the original (PDF) on 15 July 2010. Retrieved 14 March 2010.
- ^ «Annual Data Report 2016 Cystic Fibrosis Foundation Patient Registry» (PDF). p. 4. Archived from the original (PDF) on 19 June 2018. Retrieved 19 June 2018.
- ^ «Cystic Fibrosis Patient Registry Annual Data Report 2009» (PDF). Cystic Fibrosis Foundation. 2009. Archived from the original (PDF) on 5 January 2012.
- ^ Yu H, Nasr SZ, Deretic V (April 2000). «Innate lung defenses and compromised Pseudomonas aeruginosa clearance in the malnourished mouse model of respiratory infections in cystic fibrosis». Infection and Immunity. 68 (4): 2142–7. doi:10.1128/IAI.68.4.2142-2147.2000. PMC 97396. PMID 10722612.
- ^ Ratjen F, Döring G (February 2003). «Cystic fibrosis». Lancet. 361 (9358): 681–9. doi:10.1016/S0140-6736(03)12567-6. PMID 12606185. S2CID 24879334.
- ^ Rosenstein BJ, Zeitlin PL (January 1998). «Cystic fibrosis». Lancet. 351 (9098): 277–82. doi:10.1016/S0140-6736(97)09174-5. PMID 9457113. S2CID 44627706.
- ^ Schmitz TG, Goldbeck L (February 2006). «The effect of inpatient rehabilitation programmes on quality of life in patients with cystic fibrosis: a multi-center study». Health and Quality of Life Outcomes. 4: 8. doi:10.1186/1477-7525-4-8. PMC 1373610. PMID 16457728.
- ^ Hegarty M, Macdonald J, Watter P, Wilson C (July 2009). «Quality of life in young people with cystic fibrosis: effects of hospitalization, age and gender, and differences in parent/child perceptions». Child. 35 (4): 462–8. doi:10.1111/j.1365-2214.2008.00900.x. PMID 18991968.
Havermans T, Vreys M, Proesmans M, De Boeck C (January 2006). «Assessment of agreement between parents and children on health-related quality of life in children with cystic fibrosis». Child. 32 (1): 1–7. doi:10.1111/j.1365-2214.2006.00564.x. PMID 16398786. - ^ Moorcroft AJ, Dodd ME, Webb AK (1998). «Exercise limitations and training for patients with cystic fibrosis». Disability and Rehabilitation. 20 (6–7): 247–53. doi:10.3109/09638289809166735. PMID 9637933.
- ^ «Medications». Cystic Fibrosis Canada. 2011. No. 10684-5100 RR0001. Archived from the original on 4 September 2011.
- ^ Araújo FG, Novaes FC, Santos NP, Martins VC, Souza SM, Santos SE, Ribeiro-dos-Santos AK (January 2005). «Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the North of Brazil». Brazilian Journal of Medical and Biological Research. 38 (1): 11–5. doi:10.1590/S0100-879X2005000100003. PMID 15665983.
- ^ Tobias E (2011). Essential Medical Genetics. John Wiley & Sons. p. 312. ISBN 978-1-118-29370-6. Archived from the original on 17 April 2016.
- ^ «The Canadian Facts & Figures on Cystic Fibrosis». cysticfibrosis.ca. Archived from the original on 16 June 2013.
- ^ «Genetic Carrier Testing». Cystic Fibrosis Foundation. 2007. Archived from the original on 23 March 2010.
- ^ Rosenstein BJ, Cutting GR (April 1998). «The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel». The Journal of Pediatrics. 132 (4): 589–95. doi:10.1016/S0022-3476(98)70344-0. PMID 9580754.
- ^ Hamosh A, FitzSimmons SC, Macek M, Knowles MR, Rosenstein BJ, Cutting GR (February 1998). «Comparison of the clinical manifestations of cystic fibrosis in black and white patients». The Journal of Pediatrics. 132 (2): 255–9. doi:10.1016/S0022-3476(98)70441-X. PMID 9506637.
- ^ Farrell P, Joffe S, Foley L, Canny GJ, Mayne P, Rosenberg M (September 2007). «Diagnosis of cystic fibrosis in the Republic of Ireland: epidemiology and costs». Irish Medical Journal. 100 (8): 557–60. PMID 17955689. Archived from the original on 3 December 2013.
- ^ Hytönen M, Patjas M, Vento SI, Kauppi P, Malmberg H, Ylikoski J, Kere J (December 2001). «Cystic fibrosis gene mutations deltaF508 and 394delTT in patients with chronic sinusitis in Finland». Acta Oto-Laryngologica. 121 (8): 945–7. doi:10.1080/000164801317166835. PMID 11813900.
- ^ «WHO | Genes and human disease». Who.int. 7 December 2010. Archived from the original on 20 October 2012. Retrieved 23 January 2013.
- ^ Russell P (2011). Biology: the dynamic science (2nd ed.). Belmont, CA: Brooks/Cole, Cengage Learning. p. 304. ISBN 978-0-538-49372-7. Archived from the original on 17 April 2016.
- ^ «Genetic testing for cystic fibrosis Genetic Testing for Cystic Fibrosis». Consensus Development Conference Statement. National Institutes of Health. 14–16 April 1997. Archived from the original on 27 March 2009.
- ^ Rosenfeld M, Davis R, FitzSimmons S, Pepe M, Ramsey B (May 1997). «Gender gap in cystic fibrosis mortality». American Journal of Epidemiology. 145 (9): 794–803. doi:10.1093/oxfordjournals.aje.a009172. PMID 9143209.
- ^ Coakley RD, Sun H, Clunes LA, Rasmussen JE, Stackhouse JR, Okada SF, et al. (December 2008). «17beta-Estradiol inhibits Ca2+-dependent homeostasis of airway surface liquid volume in human cystic fibrosis airway epithelia». The Journal of Clinical Investigation. 118 (12): 4025–35. doi:10.1172/JCI33893. PMC 2582929. PMID 19033671.
- ^ Verma N, Bush A, Buchdahl R (October 2005). «Is there still a gender gap in cystic fibrosis?». Chest. 128 (4): 2824–34. doi:10.1378/chest.128.4.2824. PMID 16236961.
- ^ Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W (September 2009). «Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality». Diabetes Care. 32 (9): 1626–31. doi:10.2337/dc09-0586. PMC 2732133. PMID 19542209.
- ^ «CF worse for women ‘due to effect of estrogen’«. The Irish Times. 8 August 2010. Archived from the original on 11 August 2010.
- ^ Wennberg C, Kucinskas V (1994). «Low frequency of the delta F508 mutation in Finno-Ugrian and Baltic populations». Human Heredity. 44 (3): 169–71. doi:10.1159/000154210. PMID 8039801.
- ^ Kere J, Savilahti E, Norio R, Estivill X, de la Chapelle A (September 1990). «Cystic fibrosis mutation delta F508 in Finland: other mutations predominate». Human Genetics. 85 (4): 413–5. doi:10.1007/BF02428286. PMID 2210753. S2CID 38364780.
- ^ Wiuf C (August 2001). «Do delta F508 heterozygotes have a selective advantage?». Genetical Research. 78 (1): 41–7. CiteSeerX 10.1.1.174.7283. doi:10.1017/S0016672301005195. PMID 11556136.
- ^ Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ (October 1994). «Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model». Science. 266 (5182): 107–9. Bibcode:1994Sci…266..107G. doi:10.1126/science.7524148. PMID 7524148.
- ^ Alfonso-Sánchez MA, Pérez-Miranda AM, García-Obregón S, Peña JA (June 2010). «An evolutionary approach to the high frequency of the Delta F508 CFTR mutation in European populations». Medical Hypotheses. 74 (6): 989–92. doi:10.1016/j.mehy.2009.12.018. PMID 20110149.
- ^ Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, Evans MJ (January 1995). «The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study». The Journal of Physiology. 482 (Pt 2): 449–54. doi:10.1113/jphysiol.1995.sp020531. PMC 1157742. PMID 7714835.
- ^ Högenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, et al. (December 2000). «Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion». American Journal of Human Genetics. 67 (6): 1422–7. doi:10.1086/316911. PMC 1287919. PMID 11055897.
- ^ Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, et al. (May 1998). «Salmonella typhi uses CFTR to enter intestinal epithelial cells». Nature. 393 (6680): 79–82. Bibcode:1998Natur.393…79P. doi:10.1038/30006. PMID 9590693. S2CID 5894247.
- ^ Modiano G, Ciminelli BM, Pignatti PF (March 2007). «Cystic fibrosis and lactase persistence: a possible correlation». European Journal of Human Genetics. 15 (3): 255–9. doi:10.1038/sj.ejhg.5201749. hdl:2108/29185. PMID 17180122. S2CID 4650571.
- ^ Poolman EM, Galvani AP (February 2007). «Evaluating candidate agents of selective pressure for cystic fibrosis». Journal of the Royal Society, Interface. 4 (12): 91–8. doi:10.1098/rsif.2006.0154. PMC 2358959. PMID 17015291.
- ^ Williams N (2006). «Footprint fears for new TB threat». Current Biology. 16 (19): R821–R822. doi:10.1016/j.cub.2006.09.009. S2CID 2346727.
- ^ Tobacman JK (June 2003). «Does deficiency of arylsulfatase B have a role in cystic fibrosis?». Chest. 123 (6): 2130–9. doi:10.1378/chest.123.6.2130. PMID 12796199.
- ^ a b c Busch R (1990). «On the History of Cystic Fibrosis». Acta Universitatis Carolinae. Medica. 36 (1–4): 13–15. PMID 2130674.
- ^ Fanconi G, Uehlinger E, Knauer C (1936). «Das coeliakiesyndrom bei angeborener zysticher pankreasfibromatose und bronchiektasien». Wien. Med. Wochenschr. 86: 753–756.
- ^ Di Sant’Agnese PA, Darling RC, Perera GA, Shea E (November 1953). «Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease». Pediatrics. 12 (5): 549–563. doi:10.1542/peds.12.5.549. PMID 13111855. S2CID 42514224.
- ^ Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. (September 1989). «Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA». Science. 245 (4922): 1066–1073. Bibcode:1989Sci…245.1066R. doi:10.1126/science.2475911. PMID 2475911. S2CID 84566748.
- ^ Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. (September 1989). «Identification of the cystic fibrosis gene: chromosome walking and jumping». Science. 245 (4922): 1059–1065. Bibcode:1989Sci…245.1059R. doi:10.1126/science.2772657. PMID 2772657.
- ^ Freudenheim M (22 December 2009). «Tool in Cystic Fibrosis Fight: A Registry». The New York Times. pp. D1. Archived from the original on 24 May 2013. Retrieved 21 December 2009.
- ^ Lee TW, Southern KW, Perry LA, Penny-Dimri JC, Aslam AA (June 2016). Southern KW (ed.). «Topical cystic fibrosis transmembrane conductance regulator gene replacement for cystic fibrosis-related lung disease». The Cochrane Database of Systematic Reviews. 2016 (6): CD005599. doi:10.1002/14651858.CD005599.pub5. PMC 8682957. PMID 27314455.
- ^ Alton EW, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. (September 2015). «Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial». The Lancet. Respiratory Medicine. 3 (9): 684–691. doi:10.1016/S2213-2600(15)00245-3. PMC 4673100. PMID 26149841.
- ^ Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD (November 2002). «Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis». American Journal of Respiratory Cell and Molecular Biology. 27 (5): 619–27. doi:10.1165/rcmb.2001-0004oc. PMID 12397022. S2CID 8714332.
- ^ Tate S, Elborn S (March 2005). «Progress towards gene therapy for cystic fibrosis». Expert Opinion on Drug Delivery. 2 (2): 269–80. doi:10.1517/17425247.2.2.269. PMID 16296753. S2CID 30948229.
- ^ Online Mendelian Inheritance in Man (OMIM): CYSTIC FIBROSIS; CF — 219700
- ^ Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, et al. (December 2013). «Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients». Cell Stem Cell. 13 (6): 653–8. doi:10.1016/j.stem.2013.11.002. PMID 24315439.
- ^ Hraiech S, Brégeon F, Rolain JM (2015). «Bacteriophage-based therapy in cystic fibrosis-associated Pseudomonas aeruginosa infections: rationale and current status». Drug Design, Development and Therapy. 9: 3653–63. doi:10.2147/DDDT.S53123. PMC 4509528. PMID 26213462.
- ^ Trend S, Fonceca AM, Ditcham WG, Kicic A, Cf A (November 2017). «The potential of phage therapy in cystic fibrosis: Essential human-bacterial-phage interactions and delivery considerations for use in Pseudomonas aeruginosa-infected airways». Journal of Cystic Fibrosis. 16 (6): 663–670. doi:10.1016/j.jcf.2017.06.012. PMID 28720345.
- ^ a b Ramsey BW, Downey GP, Goss CH (May 2019). «Update in Cystic Fibrosis 2018». American Journal of Respiratory and Critical Care Medicine. 199 (10): 1188–1194. doi:10.1164/rccm.201902-0310UP. PMC 6519861. PMID 30917288. ProQuest 2230820891.
- ^ Dietz HC (August 2010). «New therapeutic approaches to mendelian disorders». The New England Journal of Medicine. 363 (9): 852–63. doi:10.1056/NEJMra0907180. PMID 20818846. S2CID 5809127. Free full text
- ^ a b Office of the Commissioner (24 October 2019). «FDA approves new breakthrough therapy for cystic fibrosis». FDA. Retrieved 13 November 2019.
- ^ «CFTR Modulator Therapies». Bethesda, Md.: Cystic Fibrosis Foundation. Retrieved 13 November 2019.
- ^ Sherrard LJ, McGrath SJ, McIlreavey L, Hatch J, Wolfgang MC, Muhlebach MS, Gilpin DF, Elborn JS, Tunney MM (February 2016). «Production of extended-spectrum beta-lactamases and the potential indirect pathogenic role of Prevotella isolates from the cystic fibrosis respiratory microbiota». Int J Antimicrob Agents. 47 (2): 140–145. doi:10.1016/j.ijantimicag.2015.12.004. PMC 4746055. PMID 26774156.
Further reading[edit]
- Egan ME, Schechter MS, Voynow JA (2020). «Cystic Fibrosis». In Kliegman RM, St Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds.). Nelson Textbook of Pediatrics. Elsevier. pp. 2282–2297. ISBN 978-0-323-56890-6.
External links[edit]
- Search GeneCards for genes involved in cystic fibrosis
- Cystic Fibrosis Mutation Database
- «Cystic Fibrosis». MedlinePlus. U.S. National Library of Medicine.
| Cystic fibrosis | |
|---|---|
| Other names | Mucoviscidosis |
|
|
| Specialty | Medical genetics, pulmonology |
| Symptoms | Difficulty breathing, coughing up mucus, poor growth, fatty stool[1] |
| Usual onset | Symptoms recognizable ~6 month[2] |
| Duration | Life long[3] |
| Causes | Genetic (autosomal recessive)[1] |
| Risk factors | Genetic |
| Diagnostic method | Sweat test, genetic testing[1] |
| Treatment | Antibiotics, pancreatic enzyme replacement, lung transplantation[1] |
| Prognosis | Life expectancy between 42 and 50 years (developed world)[4] |
| Frequency | 1 out of 3,000 (Northern European)[1] |
Cystic fibrosis (CF) is a rare[5][6] genetic disorder that affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine.[1][7] Long-term issues include difficulty breathing and coughing up mucus as a result of frequent lung infections.[1] Other signs and symptoms may include sinus infections, poor growth, fatty stool, clubbing of the fingers and toes, and infertility in most males.[1] Different people may have different degrees of symptoms.[1]
Cystic fibrosis is inherited in an autosomal recessive manner.[1] It is caused by the presence of mutations in both copies of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein.[1] Those with a single working copy are carriers and otherwise mostly healthy.[3] CFTR is involved in the production of sweat, digestive fluids, and mucus.[8] When the CFTR is not functional, secretions which are usually thin instead become thick.[9] The condition is diagnosed by a sweat test and genetic testing.[1] Screening of infants at birth takes place in some areas of the world.[1]
There is no known cure for cystic fibrosis.[3] Lung infections are treated with antibiotics which may be given intravenously, inhaled, or by mouth.[1] Sometimes, the antibiotic azithromycin is used long term.[1] Inhaled hypertonic saline and salbutamol may also be useful.[1] Lung transplantation may be an option if lung function continues to worsen.[1] Pancreatic enzyme replacement and fat-soluble vitamin supplementation are important, especially in the young.[1] Airway clearance techniques such as chest physiotherapy have some short-term benefit, but long-term effects are unclear.[10] The average life expectancy is between 42 and 50 years in the developed world.[4][11] Lung problems are responsible for death in 80% of people with cystic fibrosis.[1]
CF is most common among people of Northern European ancestry, for whom it affects about 1 out of 3,000 newborns,[1] and among which around 1 out of 25 people is a carrier.[3] It is least common in Africans and Asians, though it does occur in all races.[1] It was first recognized as a specific disease by Dorothy Andersen in 1938, with descriptions that fit the condition occurring at least as far back as 1595.[7] The name «cystic fibrosis» refers to the characteristic fibrosis and cysts that form within the pancreas.[7][12]
Signs and symptoms[edit]
Health problems associated with cystic fibrosis
Cystic fibrosis typically manifests early in life. Newborns and infants with cystic fibrosis tend to have frequent, large, greasy stools (a result of malabsorption) and are underweight for their age.[13] 15–20% of newborns have their small intestine blocked by meconium, often requiring surgery to correct.[13] Newborns occasionally have neonatal jaundice due to blockage of the bile ducts.[13] Children with cystic fibrosis lose excessive salt in their sweat, and parents often notice salt crystallizing on the skin, or a salty taste when they kiss their child.[13]
The primary cause of morbidity and death in people with cystic fibrosis is progressive lung disease, which eventually leads to respiratory failure.[14] This typically begins as a prolonged respiratory infection that continues until treated with antibiotics.[14] Chronic infection of the respiratory tract is nearly universal in people with cystic fibrosis, with Pseudomonas aeruginosa, fungi, and mycobacteria all increasingly common over time.[15] Inflammation of the upper airway results in frequent runny nose and nasal obstruction. Nasal polyps are common, particularly in children and teenagers.[14] As the disease progresses, people tend to have shortness of breath, and a chronic cough that produces sputum.[14] Breathing problems make it increasingly challenging to exercise, and prolonged illness causes those affected to be underweight for their age.[14] In late adolescence or adulthood, people begin to develop severe signs of lung disease: wheezing, digital clubbing, cyanosis, coughing up blood, pulmonary heart disease, and collapsed lung (atelectasis or pneumothorax).[14]
In rare cases, cystic fibrosis can manifest itself as a coagulation disorder. Vitamin K is normally absorbed from breast milk, formula, and later, solid foods. This absorption is impaired in some CF patients. Young children are especially sensitive to vitamin K malabsorptive disorders because only a very small amount of vitamin K crosses the placenta, leaving the child with very low reserves and limited ability to absorb vitamin K from dietary sources after birth. Because clotting factors II, VII, IX, and X are vitamin K–dependent, low levels of vitamin K can result in coagulation problems. Consequently, when a child presents with unexplained bruising, a coagulation evaluation may be warranted to determine whether an underlying disease is present.[16]
Lungs and sinuses[edit]
Lung disease results from clogging of the airways due to mucus build-up, decreased mucociliary clearance, and resulting inflammation.[17][18] In later stages, changes in the architecture of the lung, such as pathology in the major airways (bronchiectasis), further exacerbate difficulties in breathing. Other signs include high blood pressure in the lung (pulmonary hypertension), heart failure, difficulties getting enough oxygen to the body (hypoxia), and respiratory failure requiring support with breathing masks, such as bilevel positive airway pressure machines or ventilators.[19] Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa are the three most common organisms causing lung infections in CF patients.[18]: 1254 In addition, opportunistic infection due to Burkholderia cepacia complex can occur, especially through transmission from patient to patient.[20]
In addition to typical bacterial infections, people with CF more commonly develop other types of lung diseases. Among these is allergic bronchopulmonary aspergillosis, in which the body’s response to the common fungus Aspergillus fumigatus causes worsening of breathing problems. Another is infection with Mycobacterium avium complex, a group of bacteria related to tuberculosis, which can cause lung damage and do not respond to common antibiotics.[21]
Mucus in the paranasal sinuses is equally thick and may also cause blockage of the sinus passages, leading to infection. This may cause facial pain, fever, nasal drainage, and headaches. Individuals with CF may develop overgrowth of the nasal tissue (nasal polyps) due to inflammation from chronic sinus infections.[22] Recurrent sinonasal polyps can occur in 10% to 25% of CF patients.[18]: 1254 These polyps can block the nasal passages and increase breathing difficulties.[23][24]
Cardiorespiratory complications are the most common causes of death (about 80%) in patients at most CF centers in the United States.[18]: 1254
Gastrointestinal[edit]
In addition, protrusion of internal rectal membranes (rectal prolapse) is more common, occurring in as many as 10% of children with CF,[18] and it is caused by increased fecal volume, malnutrition, and increased intra–abdominal pressure due to coughing.[25]
The thick mucus seen in the lungs has a counterpart in thickened secretions from the pancreas, an organ responsible for providing digestive juices that help break down food. These secretions block the exocrine movement of the digestive enzymes into the duodenum and result in irreversible damage to the pancreas, often with painful inflammation (pancreatitis).[26] The pancreatic ducts are totally plugged in more advanced cases, usually seen in older children or adolescents.[18] This causes atrophy of the exocrine glands and progressive fibrosis.[18]
Individuals with CF also have difficulties absorbing the fat-soluble vitamins A, D, E, and K.[27]
In addition to the pancreas problems, people with CF experience more heartburn,[27] intestinal blockage by intussusception, and constipation.[28] Older individuals with CF may develop distal intestinal obstruction syndrome, which occurs when feces becomes thick with mucus (inspissated) and can cause bloating, pain, and incomplete or complete bowel obstruction.[29][27]
Exocrine pancreatic insufficiency occurs in the majority (85–90%) of patients with CF.[18]: 1253 It is mainly associated with «severe» CFTR mutations, where both alleles are completely nonfunctional (e.g. ΔF508/ΔF508).[18]: 1253 It occurs in 10–15% of patients with one «severe» and one «mild» CFTR mutation where little CFTR activity still occurs, or where two «mild» CFTR mutations exist.[18]: 1253 In these milder cases, sufficient pancreatic exocrine function is still present so that enzyme supplementation is not required.[18]: 1253 Usually, no other GI complications occur in pancreas-sufficient phenotypes, and in general, such individuals usually have excellent growth and development.[18]: 1254 Despite this, idiopathic chronic pancreatitis can occur in a subset of pancreas-sufficient individuals with CF, and is associated with recurrent abdominal pain and life-threatening complications.[18]
Thickened secretions also may cause liver problems in patients with CF. Bile secreted by the liver to aid in digestion may block the bile ducts, leading to liver damage. Impaired digestion or absorption of lipids can result in steatorrhea. Over time, this can lead to scarring and nodularity (cirrhosis). The liver fails to rid the blood of toxins and does not make important proteins, such as those responsible for blood clotting.[30][31] Liver disease is the third-most common cause of death associated with CF.[18]
Around 5–7% of people experience liver damage severe enough to cause symptoms: typically gallstones causing biliary colic.[32]
Endocrine[edit]
The pancreas contains the islets of Langerhans, which are responsible for making insulin, a hormone that helps regulate blood glucose. Damage to the pancreas can lead to loss of the islet cells, leading to a type of diabetes unique to those with the disease.[33] This cystic fibrosis-related diabetes shares characteristics of type 1 and type 2 diabetes, and is one of the principal nonpulmonary complications of CF.[34]
Vitamin D is involved in calcium and phosphate regulation. Poor uptake of vitamin D from the diet because of malabsorption can lead to the bone disease osteoporosis in which weakened bones are more susceptible to fractures.[35]
Infertility[edit]
Infertility affects both men and women. At least 97% of men with cystic fibrosis are infertile, but not sterile, and can have children with assisted reproductive techniques.[36] The main cause of infertility in men with cystic fibrosis is congenital absence of the vas deferens (which normally connects the testes to the ejaculatory ducts of the penis), but potentially also by other mechanisms such as causing no sperm, abnormally shaped sperm, and few sperm with poor motility.[37] Many men found to have congenital absence of the vas deferens during evaluation for infertility have a mild, previously undiagnosed form of CF.[38] Around 20% of women with CF have fertility difficulties due to thickened cervical mucus or malnutrition. In severe cases, malnutrition disrupts ovulation and causes a lack of menstruation.[39]
Causes[edit]
Cystic fibrosis has an autosomal recessive pattern of inheritance.
CF is caused by a mutation in the gene cystic fibrosis transmembrane conductance regulator (CFTR). The most common mutation, ΔF508, is a deletion (Δ signifying deletion) of three nucleotides that results in a loss of the amino acid phenylalanine (F) at the 508th position on the protein.[40][41] This mutation accounts for two-thirds (66–70%[18]) of CF cases worldwide and 90% of cases in the United States; however, over 1500 other mutations can produce CF.[42] Although most people have two working copies (alleles) of the CFTR gene, only one is needed to prevent cystic fibrosis. CF develops when neither allele can produce a functional CFTR protein. Thus, CF is considered an autosomal recessive disease.[43]
The CFTR gene, found at the q31.2 locus of chromosome 7, is 230,000 base pairs long, and creates a protein that is 1,480 amino acids long. More specifically, the location is between base pair 117,120,016 and 117,308,718 on the long arm of chromosome 7, region 3, band 1, subband 2, represented as 7q31.2. Structurally, the CFTR is a type of gene known as an ABC gene. The product of this gene (the CFTR protein) is a chloride ion channel important in creating sweat, digestive juices, and mucus. This protein possesses two ATP-hydrolyzing domains, which allows the protein to use energy in the form of ATP. It also contains two domains comprising six alpha helices apiece, which allow the protein to cross the cell membrane. A regulatory binding site on the protein allows activation by phosphorylation, mainly by cAMP-dependent protein kinase.[19] The carboxyl terminal of the protein is anchored to the cytoskeleton by a PDZ domain interaction.[44] The majority of CFTR in the lung’s passages is produced by rare ion-transporting cells that regulate mucus properties.[45]
In addition, the evidence is increasing that genetic modifiers besides CFTR modulate the frequency and severity of the disease. One example is mannan-binding lectin, which is involved in innate immunity by facilitating phagocytosis of microorganisms. Polymorphisms in one or both mannan-binding lectin alleles that result in lower circulating levels of the protein are associated with a threefold higher risk of end-stage lung disease, as well as an increased burden of chronic bacterial infections.[18]
Carriers[edit]
Up to one in 25 individuals of Northern European ancestry is considered a genetic carrier. The disease appears only when two of these carriers have children, as each pregnancy between them has a 25% chance of producing a child with the disease. Although only about one of every 3,000 newborns of the affected ancestry has CF, more than 900 mutations of the gene that causes CF are known. Current tests look for the most common mutations.[46]
The mutations screened by the test vary according to a person’s ethnic group or by the occurrence of CF already in the family. More than 10 million Americans, including one in 25 white Americans, are carriers of one mutation of the CF gene. CF is present in other races, though not as frequently as in white individuals. About one in 46 Hispanic Americans, one in 65 African Americans, and one in 90 Asian Americans carry a mutation of the CF gene.[46]
Pathophysiology[edit]
The CFTR protein is a channel protein that controls the flow of H2O and Cl− ions in and out of cells inside the lungs. When the CFTR protein is working correctly, ions freely flow in and out of the cells. However, when the CFTR protein is malfunctioning, these ions cannot flow out of the cell due to a blocked channel. This causes cystic fibrosis, characterized by the buildup of thick mucus in the lungs.
Several mutations in the CFTR gene can occur, and different mutations cause different defects in the CFTR protein, sometimes causing a milder or more severe disease. These protein defects are also targets for drugs which can sometimes restore their function. ΔF508-CFTR gene mutation, which occurs in >90% of patients in the U.S., creates a protein that does not fold normally and is not appropriately transported to the cell membrane, resulting in its degradation.[47]
Other mutations result in proteins that are too short (truncated) because production is ended prematurely. Other mutations produce proteins that do not use energy (in the form of ATP) normally, do not allow chloride, iodide, and thiocyanate to cross the membrane appropriately,[48] and degrade at a faster rate than normal. Mutations may also lead to fewer copies of the CFTR protein being produced.[19]
The protein created by this gene is anchored to the outer membrane of cells in the sweat glands, lungs, pancreas, and all other remaining exocrine glands in the body.
The protein spans this membrane and acts as a channel connecting the inner part of the cell (cytoplasm) to the surrounding fluid. This channel is primarily responsible for controlling the movement of halide anions from inside to outside of the cell; however, in the sweat ducts, it facilitates the movement of chloride from the sweat duct into the cytoplasm. When the CFTR protein does not resorb ions in sweat ducts, chloride and thiocyanate[49] released from sweat glands are trapped inside the ducts and pumped to the skin.
Additionally hypothiocyanite, OSCN, cannot be produced by the immune defense system.[50][51] Because chloride is negatively charged, this modifies the electrical potential inside and outside the cell that normally causes cations to cross into the cell. Sodium is the most common cation in the extracellular space. The excess chloride within sweat ducts prevents sodium resorption by epithelial sodium channels and the combination of sodium and chloride creates the salt, which is lost in high amounts in the sweat of individuals with CF. This lost salt forms the basis for the sweat test.[19]
Most of the damage in CF is due to blockage of the narrow passages of affected organs with thickened secretions. These blockages lead to remodeling and infection in the lung, damage by accumulated digestive enzymes in the pancreas, blockage of the intestines by thick feces, etc. Several theories have been posited on how the defects in the protein and cellular function cause the clinical effects. The most current theory suggests that defective ion transport leads to dehydration in the airway epithelia, thickening mucus.[52] In airway epithelial cells, the cilia exist in between the cell’s apical surface and mucus in a layer known as airway surface liquid (ASL). The flow of ions from the cell and into this layer is determined by ion channels such as CFTR. CFTR not only allows chloride ions to be drawn from the cell and into the ASL, but it also regulates another channel called ENac, which allows sodium ions to leave the ASL and enter the respiratory epithelium. CFTR normally inhibits this channel, but if the CFTR is defective, then sodium flows freely from the ASL and into the cell.[citation needed]
As water follows sodium, the depth of ASL will be depleted and the cilia will be left in the mucous layer.[53] As cilia cannot effectively move in a thick, viscous environment, mucociliary clearance is deficient and a buildup of mucus occurs, clogging small airways.[54] The accumulation of more viscous, nutrient-rich mucus in the lungs allows bacteria to hide from the body’s immune system, causing repeated respiratory infections. The presence of the same CFTR proteins in the pancreatic duct and sweat glands in the skin also cause symptoms in these systems.[citation needed]
Chronic infections[edit]
The lungs of individuals with cystic fibrosis are colonized and infected by bacteria from an early age. These bacteria, which often spread among individuals with CF, thrive in the altered mucus, which collects in the small airways of the lungs. This mucus leads to the formation of bacterial microenvironments known as biofilms that are difficult for immune cells and antibiotics to penetrate. Viscous secretions and persistent respiratory infections repeatedly damage the lung by gradually remodeling the airways, which makes infection even more difficult to eradicate.[55] The natural history of CF lung infections and airway remodeling is poorly understood, largely due to the immense spatial and temporal heterogeneity both within and between the microbiomes of CF patients.[56]
Over time, both the types of bacteria and their individual characteristics change in individuals with CF. In the initial stage, common bacteria such as S. aureus and H. influenzae colonize and infect the lungs.[18] Eventually, Pseudomonas aeruginosa (and sometimes Burkholderia cepacia) dominates. By 18 years of age, 80% of patients with classic CF harbor P. aeruginosa, and 3.5% harbor B. cepacia.[18] Once within the lungs, these bacteria adapt to the environment and develop resistance to commonly used antibiotics. Pseudomonas can develop special characteristics that allow the formation of large colonies, known as «mucoid» Pseudomonas, which are rarely seen in people who do not have CF.[55] Scientific evidence suggests the interleukin 17 pathway plays a key role in resistance and modulation of the inflammatory response during P. aeruginosa infection in CF.[57] In particular, interleukin 17-mediated immunity plays a double-edged activity during chronic airways infection; on one side, it contributes to the control of P. aeruginosa burden, while on the other, it propagates exacerbated pulmonary neutrophilia and tissue remodeling.[57]
Infection can spread by passing between different individuals with CF.[58] In the past, people with CF often participated in summer «CF camps» and other recreational gatherings.[59][60] Hospitals grouped patients with CF into common areas and routine equipment (such as nebulizers)[61] was not sterilized between individual patients.[62] This led to transmission of more dangerous strains of bacteria among groups of patients. As a result, individuals with CF are now routinely isolated from one another in the healthcare setting, and healthcare providers are encouraged to wear gowns and gloves when examining patients with CF to limit the spread of virulent bacterial strains.[63]
CF patients may also have their airways chronically colonized by filamentous fungi (such as Aspergillus fumigatus, Scedosporium apiospermum, Aspergillus terreus) and/or yeasts (such as Candida albicans); other filamentous fungi less commonly isolated include Aspergillus flavus and Aspergillus nidulans (occur transiently in CF respiratory secretions) and Exophiala dermatitidis and Scedosporium prolificans (chronic airway-colonizers); some filamentous fungi such as Penicillium emersonii and Acrophialophora fusispora are encountered in patients almost exclusively in the context of CF.[64] Defective mucociliary clearance characterizing CF is associated with local immunological disorders. In addition, the prolonged therapy with antibiotics and the use of corticosteroid treatments may also facilitate fungal growth. Although the clinical relevance of the fungal airway colonization is still a matter of debate, filamentous fungi may contribute to the local inflammatory response and therefore to the progressive deterioration of the lung function, as often happens with allergic bronchopulmonary aspergillosis – the most common fungal disease in the context of CF, involving a Th2-driven immune response to Aspergillus species.[64][65]
Diagnosis[edit]
The location of the CFTR gene on chromosome 7
In many localities all newborns are screened for cystic fibrosis within the first few days of life, typically by blood test for high levels of immunoreactive trypsinogen.[66] Newborns with positive tests or those who are otherwise suspected of having cystic fibrosis based on symptoms or family history, then undergo a sweat test. An electric current is used to drive pilocarpine into the skin, stimulating sweating. The sweat is collected and analyzed for salt levels. Having unusually high levels of chloride in the sweat suggests CFTR is dysfunctional; the person is then diagnosed with cystic fibrosis.[67][note 1] Genetic testing is also available to identify the CFTR mutations typically associated with cystic fibrosis. Many laboratories can test for the 30–96 most common CFTR mutations, which can identify over 90% of people with cystic fibrosis.[67]
People with CF have less thiocyanate and hypothiocyanite in their saliva[69] and mucus (Banfi et al.). In the case of milder forms of CF, transepithelial potential difference measurements can be helpful. CF can also be diagnosed by identification of mutations in the CFTR gene.[70]
In many cases, a parent makes the diagnosis because the infant tastes salty.[18] Immunoreactive trypsinogen levels can be increased in individuals who have a single mutated copy of the CFTR gene (carriers) or, in rare instances, in individuals with two normal copies of the CFTR gene. Due to these false positives, CF screening in newborns can be controversial.[71][72]
By 2010 every US state had instituted newborn screening programs [73] and as of 2016, 21 European countries had programs in at least some regions.[74]
Prenatal[edit]
Women who are pregnant or couples planning a pregnancy can have themselves tested for the CFTR gene mutations to determine the risk that their child will be born with CF. Testing is typically performed first on one or both parents and, if the risk of CF is high, testing on the fetus is performed. The American College of Obstetricians and Gynecologists recommends all people thinking of becoming pregnant be tested to see if they are a carrier.[75]
Because development of CF in the fetus requires each parent to pass on a mutated copy of the CFTR gene and because CF testing is expensive, testing is often performed initially on one parent. If testing shows that parent is a CFTR gene mutation carrier, the other parent is tested to calculate the risk that their children will have CF. CF can result from more than a thousand different mutations.[43] As of 2016, typically only the most common mutations are tested for, such as ΔF508[43] Most commercially available tests look for 32 or fewer different mutations. If a family has a known uncommon mutation, specific screening for that mutation can be performed. Because not all known mutations are found on current tests, a negative screen does not guarantee that a child will not have CF.[76]
During pregnancy, testing can be performed on the placenta (chorionic villus sampling) or the fluid around the fetus (amniocentesis). However, chorionic villus sampling has a risk of fetal death of one in 100 and amniocentesis of one in 200;[77] a recent study has indicated this may be much lower, about one in 1,600.[78]
Economically, for carrier couples of cystic fibrosis, when comparing preimplantation genetic diagnosis (PGD) with natural conception (NC) followed by prenatal testing and abortion of affected pregnancies, PGD provides net economic benefits up to a maternal age around 40 years, after which NC, prenatal testing, and abortion have higher economic benefit.[79]
Management[edit]
While no cures for CF are known, several treatment methods are used. The management of CF has improved significantly over the past 70 years. While infants born with it 70 years ago would have been unlikely to live beyond their first year, infants today are likely to live well into adulthood. Recent advances in the treatment of cystic fibrosis have meant that individuals with cystic fibrosis can live a fuller life less encumbered by their condition. The cornerstones of management are the proactive treatment of airway infection, and encouragement of good nutrition and an active lifestyle. Pulmonary rehabilitation as a management of CF continues throughout a person’s life, and is aimed at maximizing organ function, and therefore the quality of life. Occupational therapists use energy conservation techniques (ECT) in the rehabilitation process for patients with Cystic Fibrosis.[80] Examples of energy conservation techniques are ergonomic principles, pursed lip breathing, and diaphragmatic breathing.[81] Patients with CF tend to have fatigue and dyspnoea due to chronic pulmonary infections, so reducing the amount of energy spent during activities can help patients feel better and gain more independence.[80] At best, current treatments delay the decline in organ function. Because of the wide variation in disease symptoms, treatment typically occurs at specialist multidisciplinary centers and is tailored to the individual. Targets for therapy are the lungs, gastrointestinal tract (including pancreatic enzyme supplements), the reproductive organs (including assisted reproductive technology), and psychological support.[82]
The most consistent aspect of therapy in CF is limiting and treating the lung damage caused by thick mucus and infection, with the goal of maintaining quality of life. Intravenous, inhaled, and oral antibiotics are used to treat chronic and acute infections. Mechanical devices and inhalation medications are used to alter and clear the thickened mucus. These therapies, while effective, can be extremely time-consuming. Oxygen therapy at home is recommended in those with significant low oxygen levels.[83] Many people with CF use probiotics, which are thought to be able to correct intestinal dysbiosis and inflammation, but the clinical trial evidence regarding the effectiveness of probiotics for reducing pulmonary exacerbations in people with CF is uncertain.[84]
Antibiotics[edit]
Many people with CF are on one or more antibiotics at all times, even when healthy, to prophylactically suppress infection. Antibiotics are absolutely necessary whenever pneumonia is suspected or a noticeable decline in lung function is seen, and are usually chosen based on the results of a sputum analysis and the person’s past response. This prolonged therapy often necessitates hospitalization and insertion of a more permanent IV such as a peripherally inserted central catheter or Port-a-Cath. Inhaled therapy with antibiotics such as tobramycin, colistin, and aztreonam is often given for months at a time to improve lung function by impeding the growth of colonized bacteria.[85][86][87] Inhaled antibiotic therapy helps lung function by fighting infection, but also has significant drawbacks such as development of antibiotic resistance, tinnitus, and changes in the voice.[88] Inhaled levofloxacin may be used to treat Pseudomonas aeruginosa in people with cystic fibrosis who are infected.[89] The early management of Pseudomonas aeruginosa infection is easier and better, using nebulised antibiotics with or without oral antibiotics may sustain its eradication up to two years.[90] When choosing antibiotics to treat CF patients with lung infections caused by Pseudomonas aeruginosa in people with cystic fibrosis, it is still unclear whether the choice of antibiotics should be based on the results of testing antibiotics separately (one at a time) or in combination with each other.[91]
Antibiotics by mouth such as ciprofloxacin or azithromycin are given to help prevent infection or to control ongoing infection.[92] The aminoglycoside antibiotics (e.g. tobramycin) used can cause hearing loss, damage to the balance system in the inner ear or kidney failure with long-term use.[93] To prevent these side-effects, the amount of antibiotics in the blood is routinely measured and adjusted accordingly.[94]
All these factors related to the antibiotics use, the chronicity of the disease, and the emergence of resistant bacteria demand more exploration for different strategies such as antibiotic adjuvant therapy.[95] Currently, no reliable clinical trial evidence shows the effectiveness of antibiotics for pulmonary exacerbations in people with cystic fibrosis and Burkholderia cepacia complex[96] or for the use of antibiotics to treat nontuberculous mycobacteria in people with CF.[97]
Other medication[edit]
Aerosolized medications that help loosen secretions include dornase alfa and hypertonic saline.[98] Dornase is a recombinant human deoxyribonuclease, which breaks down DNA in the sputum, thus decreasing its viscosity.[99] Dornase alpha improves lung function and probably decreases the risk of exacerbations but there is insufficient evidence to know if it is more or less effective than other similar medications.[100] Dornase alpha may improve lung function, however there is no strong evidence that it is better than other hyperosmolar therapies.[100]
Denufosol, an investigational drug, opens an alternative chloride channel, helping to liquefy mucus.[101] Whether inhaled corticosteroids are useful is unclear, but stopping inhaled corticosteroid therapy is safe.[102] There is weak evidence that corticosteroid treatment may cause harm by interfering with growth.[102] Pneumococcal vaccination has not been studied as of 2014.[103] As of 2014, there is no clear evidence from randomized controlled trials that the influenza vaccine is beneficial for people with cystic fibrosis.[104]
Ivacaftor is a medication taken by mouth for the treatment of CF due to a number of specific mutations responsive to ivacaftor-induced CFTR protein enhancement.[105][106] It improves lung function by about 10%; however, as of 2014 it is expensive.[105] The first year it was on the market, the list price was over $300,000 per year in the United States.[105][needs update] In July 2015, the U.S. Food and Drug Administration approved lumacaftor/ivacaftor.[107] In 2018, the FDA approved the combination ivacaftor/tezacaftor; the manufacturer announced a list price of $292,000 per year.[108] Tezacaftor helps move the CFTR protein to the correct position on the cell surface, and is designed to treat people with the F508del mutation.[109]
In 2019, the combination drug elexacaftor/ivacaftor/tezacaftor marketed as Trikafta in the United States, was approved for CF patients over the age of 12.[110][111] In 2021, this was extended to include patients over the age of 6.[112] In Europe this drug was approved in 2020 and marketed as Kaftrio.[113] It is used in those that have a f508del mutation, which occurs in about 90% of patients with cystic fibrosis.[110][114] According to the Cystic Fibrosis Foundation, «this medicine represents the single greatest therapeutic advancement in the history of CF, offering a treatment for the underlying cause of the disease that could eventually bring modulator therapy to 90 percent of people with CF.»[115] In a clinical trial, participants who were administered the combination drug experienced a subsequent 63% decrease in pulmonary exacerbations and a 41.8 mmol/L decrease in sweat chloride concentration.[116] By mitigating a repertoire of symptoms associated with cystic fibrosis, the combination drug significantly improved quality-of-life metrics among patients with the disease as well.[116][115] The combination drug is also known to interact with CYP3A inducers, such as carbamazepine used in the treatment of bipolar disorder, causing elexafaftor/ivacaftor/tezacaftor to circulate in the body at decreased concentrations. As such, concomitant use is not recommended.[117] The list price in the US is going to be $311,000 per year;[118] however, insurance may cover much of the cost of the drug.[119]
Ursodeoxycholic acid, a bile salt, has been used, however there is insufficient data to show if it is effective.[120]
Nutrient supplementation[edit]
It is uncertain whether vitamin A or beta-carotene supplementation have any effect on eye and skin problems caused by vitamin A deficiency.[121]
There is no strong evidence that people with cystic fibrosis can prevent osteoporosis by increasing their intake of vitamin D.[122]
For people with vitamin E deficiency and cystic fibrosis, there is evidence that vitamin E supplementation may improve vitamin E levels, although it is still uncertain what effect supplementation has on vitamin E‐specific deficiency disorders or on lung function.[123]
Robust evidence regarding the effects of vitamin K supplementation in people with cystic fibrosis is lacking as of 2020.[124]
Various studies have examined the effects of omega-3 fatty acid supplementation for people with cystic fibrosis but the evidence is uncertain whether it has any benefits or adverse effects.[125]
Procedures[edit]
Several mechanical techniques are used to dislodge sputum and encourage its expectoration. One technique good for short-term airway clearance is chest physiotherapy where a respiratory therapist percusses an individual’s chest by hand several times a day, to loosen up secretions. This «percussive effect» can be administered also through specific devices that use chest wall oscillation or intrapulmonary percussive ventilator. Other methods such as biphasic cuirass ventilation, and associated clearance mode available in such devices, integrate a cough assistance phase, as well as a vibration phase for dislodging secretions. These are portable and adapted for home use.[10]
Another technique is positive expiratory pressure physiotherapy that consists of providing a back pressure to the airways during expiration. This effect is provided by devices that consists of a mask or a mouthpiece in which a resistance is applied only on the expiration phase.[126] Operating principles of this technique seems to be the increase of gas pressure behind mucus through collateral ventilation along with a temporary increase in functional residual capacity preventing the early collapse of small airways during exhalation.[127][128]
As lung disease worsens, mechanical breathing support may become necessary. Individuals with CF may need to wear special masks at night to help push air into their lungs. These machines, known as bilevel positive airway pressure (BiPAP) ventilators, help prevent low blood oxygen levels during sleep. Non-invasive ventilators may be used during physical therapy to improve sputum clearance.[129] It is not known if this type of therapy has an impact on pulmonary exacerbations or disease progression.[129] It is not known what role non-invasive ventilation therapy has for improving exercise capacity in people with cystic fibrosis.[129] However, the authors noted that «non‐invasive ventilation may be a useful adjunct to other airway clearance techniques, particularly in people with cystic fibrosis who have difficulty expectorating sputum.»[130] During severe illness, a tube may be placed in the throat (a procedure known as a tracheostomy) to enable breathing supported by a ventilator.[131][132]
For children, preliminary studies show massage therapy may help people and their families’ quality of life.[133]
Some lung infections require surgical removal of the infected part of the lung. If this is necessary many times, lung function is severely reduced.[134] The most effective treatment options for people with CF who have spontaneous or recurrent pneumothoraces is not clear.[135]
Transplantation[edit]
Lung transplantation may become necessary for individuals with CF as lung function and exercise tolerance decline. Although single lung transplantation is possible in other diseases, individuals with CF must have both lungs replaced because the remaining lung might contain bacteria that could infect the transplanted lung. A pancreatic or liver transplant may be performed at the same time to alleviate liver disease and/or diabetes.[136] Lung transplantation is considered when lung function declines to the point where assistance from mechanical devices is required or someone’s survival is threatened.[137] According to Merck Manual, «bilateral lung transplantation for severe lung disease is becoming more routine and more successful with experience and improved techniques. Among adults with CF, median survival posttransplant is about 9 years.»[138]
Other aspects[edit]
Intracytoplasmic sperm injection can be used to provide fertility for men with cystic fibrosis.
Newborns with intestinal obstruction typically require surgery, whereas adults with distal intestinal obstruction syndrome typically do not. Treatment of pancreatic insufficiency by replacement of missing digestive enzymes allows the duodenum to properly absorb nutrients and vitamins that would otherwise be lost in the feces. However, the best dosage and form of pancreatic enzyme replacement is unclear, as are the risks and long-term effectiveness of this treatment.[139]
So far, no large-scale research involving the incidence of atherosclerosis and coronary heart disease in adults with cystic fibrosis has been conducted. This is likely because the vast majority of people with cystic fibrosis do not live long enough to develop clinically significant atherosclerosis or coronary heart disease.[140]
Diabetes is the most common nonpulmonary complication of CF. It mixes features of type 1 and type 2 diabetes, and is recognized as a distinct entity, cystic fibrosis-related diabetes.[34][141] While oral antidiabetic drugs are sometimes used, the recommended treatment is the use of insulin injections or an insulin pump,[142] and, unlike in type 1 and 2 diabetes, dietary restrictions are not recommended.[34] While Stenotrophomonas maltophilia is relatively common in people with cystic fibrosis, the evidence about the effectiveness of antibiotics for S. maltophilia is uncertain.[143]
Bisphosphonates taken by mouth or intravenously can be used to improve the bone mineral density in people with cystic fibrosis.[144][needs update] When taking bisphosphates intravenously, adverse effects such as pain and flu-like symptoms can be an issue.[144] The adverse effects of bisphosphates taken by mouth on the gastrointestinal tract are not known.[144]
Poor growth may be avoided by insertion of a feeding tube for increasing food energy through supplemental feeds or by administration of injected growth hormone.[145]
Sinus infections are treated by prolonged courses of antibiotics. The development of nasal polyps or other chronic changes within the nasal passages may severely limit airflow through the nose, and over time reduce the person’s sense of smell. Sinus surgery is often used to alleviate nasal obstruction and to limit further infections. Nasal steroids such as fluticasone propionate are used to decrease nasal inflammation.[146]
Female infertility may be overcome by assisted reproduction technology, particularly embryo transfer techniques. Male infertility caused by absence of the vas deferens may be overcome with testicular sperm extraction, collecting sperm cells directly from the testicles. If the collected sample contains too few sperm cells to likely have a spontaneous fertilization, intracytoplasmic sperm injection can be performed.[147] Third party reproduction is also a possibility for women with CF. Whether taking antioxidants affects outcomes is unclear.[148]
Physical exercise is usually part of outpatient care for people with cystic fibrosis.[149] Aerobic exercise seems to be beneficial for aerobic exercise capacity, lung function and health-related quality of life; however, the quality of the evidence was poor.[149]
Due to the use of aminoglycoside antibiotics, ototoxicity is common. Symptoms may include «tinnitus, hearing loss, hyperacusis, aural fullness, dizziness, and vertigo».[150]
Gastrointestinal[edit]
Problems with the gastrointestinal system including constipation and obstruction of the gastrointestinal tract including distal intestinal obstruction syndrome are frequent complications for people with cystic fibrosis.[29] Treatment of gastrointestinal problems is required in order to prevent a complete obstruction, reduce other CF symptoms, and improve the quality of life.[29] While stool softeners, laxatives, and prokinetics (GI-focused treatments) are often suggested, there is no clear consensus from experts at to which approach is the best and comes with the least risks.[29] Mucolytics or systemic treatments aimed at dysfunctional CFTR are also sometimes suggested to improve symptoms.[151]
Prognosis[edit]
The prognosis for cystic fibrosis has improved due to earlier diagnosis through screening and better treatment and access to health care. In 1959, the median age of survival of children with CF in the United States was six months.[152]
In 2010, survival is estimated to be 37 years for women and 40 for men.[153] In Canada, median survival increased from 24 years in 1982 to 47.7 in 2007.[154] In the United States those born with CF in 2016 have an predicted life expectancy of 47.7 when cared for in specialty clinics.[155]
In the US, of those with CF who are more than 18 years old as of 2009, 92% had graduated from high school, 67% had at least some college education, 15% were disabled, 9% were unemployed, 56% were single, and 39% were married or living with a partner.[156]
Quality of life[edit]
Chronic illnesses can be difficult to manage. CF is a chronic illness that affects the «digestive and respiratory tracts resulting in generalized malnutrition and chronic respiratory infections».[157] The thick secretions clog the airways in the lungs, which often cause inflammation and severe lung infections.[158][159] If it is compromised, it affects the quality of life of someone with CF and their ability to complete such tasks as everyday chores.[citation needed]
According to Schmitz and Goldbeck (2006), CF significantly increases emotional stress on both the individual and the family, «and the necessary time-consuming daily treatment routine may have further negative effects on quality of life».[160] However, Havermans and colleagues (2006) have established that young outpatients with CF who have participated in the Cystic Fibrosis Questionnaire-Revised «rated some quality of life domains higher than did their parents».[161] Consequently, outpatients with CF have a more positive outlook for themselves. As Merck Manual notes, «with appropriate support, most patients can make an age-appropriate adjustment at home and school. Despite myriad problems, the educational, occupational, and marital successes of patients are impressive.»[138]
Furthermore, there are many ways to enhance the quality of life in CF patients. Exercise is promoted to increase lung function. Integrating an exercise regimen into the CF patient’s daily routine can significantly improve quality of life.[162] No definitive cure for CF is known, but diverse medications are used, such as mucolytics, bronchodilators, steroids, and antibiotics, that have the purpose of loosening mucus, expanding airways, decreasing inflammation, and fighting lung infections, respectively.[163]
Epidemiology[edit]
| Mutation | Frequency worldwide[164] |
|---|---|
| ΔF508 | 66–70%[18] |
| G542X | 2.4% |
| G551D | 1.6% |
| N1303K | 1.3% |
| W1282X | 1.2% |
| All others | 27.5% |
Cystic fibrosis is the most common life-limiting autosomal recessive disease among people of European heritage.[165] In the United States, about 30,000 individuals have CF; most are diagnosed by six months of age. In Canada, about 4,000 people have CF.[166] Around 1 in 25 people of European descent, and one in 30 of white Americans,[167] is a carrier of a CF mutation. Although CF is less common in these groups, roughly one in 46 Hispanics, one in 65 Africans, and one in 90 Asians carry at least one abnormal CFTR gene.[168][169] Ireland has the world’s highest prevalence of CF, at one in 1353.[170]
Although technically a rare disease, CF is ranked as one of the most widespread life-shortening genetic diseases. It is most common among nations in the Western world. An exception is Finland, where only one in 80 people carries a CF mutation.[171] The World Health Organization states, «In the European Union, one in 2000–3000 newborns is found to be affected by CF».[172] In the United States, one in 3,500 children is born with CF.[173] In 1997, about one in 3,300 white children in the United States was born with CF. In contrast, only one in 15,000 African American children have it, and in Asian Americans, the rate was even lower at one in 32,000.[174]
Cystic fibrosis is diagnosed equally in males and females. For reasons that remain unclear, data have shown that males tend to have a longer life expectancy than females,[175][176] though recent studies suggest this gender gap may no longer exist, perhaps due to improvements in health care facilities.[177][178] A recent study from Ireland identified a link between the female hormone estrogen and worse outcomes in CF.[179]
The distribution of CF alleles varies among populations. The frequency of ΔF508 carriers has been estimated at one in 200 in northern Sweden, one in 143 in Lithuanians, and one in 38 in Denmark. No ΔF508 carriers were found among 171 Finns and 151 Saami people.[180] ΔF508 does occur in Finland, but it is a minority allele there. CF is known to occur in only 20 families (pedigrees) in Finland.[181]
Evolution[edit]
The ΔF508 mutation is estimated to be up to 52,000 years old.[182] Numerous hypotheses have been advanced as to why such a lethal mutation has persisted and spread in the human population. Other common autosomal recessive diseases such as sickle-cell anemia have been found to protect carriers from other diseases, an evolutionary trade-off known as heterozygote advantage. Resistance to the following have all been proposed as possible sources of heterozygote advantage:
- Cholera: With the discovery that cholera toxin requires normal host CFTR proteins to function properly, it was hypothesized that carriers of mutant CFTR genes benefited from resistance to cholera and other causes of diarrhea.[183][184] Further studies have not confirmed this hypothesis.[185][186]
- Typhoid: Normal CFTR proteins are also essential for the entry of Salmonella Typhi into cells,[187] suggesting that carriers of mutant CFTR genes might be resistant to typhoid fever. No in vivo study has yet confirmed this. In both cases, the low level of cystic fibrosis outside of Europe, in places where both cholera and typhoid fever are endemic, is not immediately explicable.
- Diarrhea: The prevalence of CF in Europe might be connected with the development of cattle domestication. In this hypothesis, carriers of a single mutant CFTR had some protection from diarrhea caused by lactose intolerance, before the mutations that created lactose tolerance appeared.[188]
- Tuberculosis: Another possible explanation is that carriers of the gene could have some resistance to tuberculosis.[189][190] This hypothesis is based on the thesis that CFTR gene mutation carriers have insufficient action in one of their enzymes – arylsulphatase — which is necessary for Mycobacterium tuberculosis virulence. As M. tuberculosis would use its host’s sources to affect the individual, and due to the lack of enzyme it could not presents its virulence, being a carrier of CFTR mutation could provide resistance against tuberculosis.[191]
History[edit]
CF is supposed to have appeared about 3,000 BC because of migration of peoples, gene mutations, and new conditions in nourishment.[192] Although the entire clinical spectrum of CF was not recognized until the 1930s, certain aspects of CF were identified much earlier. Indeed, literature from Germany and Switzerland in the 18th century warned «Wehe dem Kind, das beim Kuß auf die Stirn salzig schmeckt, es ist verhext und muss bald sterben» («Woe to the child who tastes salty from a kiss on the brow, for he is cursed and soon must die»), recognizing the association between the salt loss in CF and illness.[192]
In the 19th century, Carl von Rokitansky described a case of fetal death with meconium peritonitis, a complication of meconium ileus associated with CF. Meconium ileus was first described in 1905 by Karl Landsteiner.[192] In 1936, Guido Fanconi described a connection between celiac disease, cystic fibrosis of the pancreas, and bronchiectasis.[193]
In 1938, Dorothy Hansine Andersen published an article, «Cystic Fibrosis of the Pancreas and Its Relation to Celiac Disease: a Clinical and Pathological Study», in the American Journal of Diseases of Children. She was the first to describe the characteristic cystic fibrosis of the pancreas and to correlate it with the lung and intestinal disease prominent in CF.[12] She also first hypothesized that CF was a recessive disease and first used pancreatic enzyme replacement to treat affected children. In 1952, Paul di Sant’Agnese discovered abnormalities in sweat electrolytes; a sweat test was developed and improved over the next decade.[194]
The first linkage between CF and another marker (paraoxonase) was found in 1985 by Hans Eiberg, indicating that only one locus exists for CF. In 1988, the first mutation for CF, ΔF508, was discovered by Francis Collins, Lap-Chee Tsui, and John R. Riordan on the seventh chromosome. Subsequent research has found over 1,000 different mutations that cause CF.[citation needed]
Because mutations in the CFTR gene are typically small, classical genetics techniques had been unable to accurately pinpoint the mutated gene.[195] Using protein markers, gene-linkage studies were able to map the mutation to chromosome 7. Chromosome walking and chromosome jumping techniques were then used to identify and sequence the gene.[196] In 1989, Lap-Chee Tsui led a team of researchers at the Hospital for Sick Children in Toronto that discovered the gene responsible for CF. CF represents a classic example of how a human genetic disorder was elucidated strictly by the process of forward genetics.[citation needed]
Research[edit]
People with CF may be listed in a disease registry that allows researchers and doctors to track health results and identify candidates for clinical trials.[197]
Gene therapy[edit]
Gene therapy has been explored as a potential cure for CF. Results from clinical trials have shown limited success as of 2016, and using gene therapy as routine therapy is not suggested.[198] A small study published in 2015 found a small benefit.[199]
The focus of much CF gene therapy research is aimed at trying to place a normal copy of the CFTR gene into affected cells. Transferring the normal CFTR gene into the affected epithelium cells would result in the production of functional CFTR protein in all target cells, without adverse reactions or an inflammation response. To prevent the lung manifestations of CF, only 5–10% the normal amount of CFTR gene expression is needed.[200] Multiple approaches have been tested for gene transfer, such as liposomes and viral vectors in animal models and clinical trials. However, both methods were found to be relatively inefficient treatment options,[201] mainly because very few cells take up the vector and express the gene, so the treatment has little effect. Additionally, problems have been noted in cDNA recombination, such that the gene introduced by the treatment is rendered unusable.[202] There has been a functional repair in culture of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients.[203]
Phage therapy[edit]
Phage therapy is being studied for multidrug resistant bacteria in people with CF.[204][205]
Gene modulators[edit]
A number of small molecules that aim at compensating various mutations of the CFTR gene are under development. CFTR modulator therapies have been used in place of other types of genetic therapies. These therapies focus on the expression of a genetic mutation instead of the mutated gene itself. Modulators are split into two classes: potentiators and correctors. Potentiators act on the CFTR ion channels that are embedded in the cell membrane, and these types of drugs help open up the channel to allow transmembrane flow. Correctors are meant to assist in the transportation of nascent proteins, a protein that is formed by ribosomes before it is morphed into a specific shape, to the cell surface to be implemented into the cell membrane.[206]
Most target the transcription stage of genetic expression. One approach has been to try and develop medication that get the ribosome to overcome the stop codon and produce a full-length CFTR protein. About 10% of CF results from a premature stop codon in the DNA, leading to early termination of protein synthesis and truncated proteins. These drugs target nonsense mutations such as G542X, which consists of the amino acid glycine in position 542 being replaced by a stop codon. Aminoglycoside antibiotics interfere with protein synthesis and error-correction. In some cases, they can cause the cell to overcome a premature stop codon by inserting a random amino acid, thereby allowing expression of a full-length protein. Future research for these modulators is focused on the cellular targets that can be effected by a change in a gene’s expression. Otherwise, genetic therapy will be used as a treatment when modulator therapies do not work given that 10% of people with cystic fibrosis are not affected by these drugs.[207]
Elexacaftor/ivacaftor/tezacaftor was approved in the United States in 2019 for cystic fibrosis.[208] This combination of previously developed medicines is able to treat up to 90% of people with cystic fibrosis.[206][208] This medications restores some effectiveness of the CFTR protein so that it can work as an ion channel on the cell’s surface.[209]
Ecological therapy[edit]
It has previously been shown that inter-species interactions are an important contributor to the pathology of CF lung infections. Examples include the production of antibiotic degrading enzymes such as β-lactamases and the production of metabolic by-products such as short-chain fatty acids (SCFAs) by anaerobic species, which can enhance the pathogenicity of traditional pathogens such as Pseudomonas aeruginosa.[210] Due to this, it has been suggested that the direct alteration of CF microbial community composition and metabolic function would provide an alternative to traditional antibiotic therapies.[56]
Society and culture[edit]
- Salt in My Soul: An Unfinished Life a posthumous memoir by Mallory Smith, a Californian with CF
- Sick: The Life and Death of Bob Flanagan, Supermasochist, a 1997 documentary film
- 65 Redroses, a 2009 documentary film
- Breathing for a Living, a memoir by Laura Rothenberg
- Every Breath I Take, Surviving and Thriving With Cystic Fibrosis, book by Claire Wineland
- Five Feet Apart, a 2019 romantic drama film starring Cole Sprouse and Haley Lu Richardson
- Orla Tinsley: Warrior, a 2018 documentary film about CF campaigner Orla Tinsley
- The performance art of Martin O’Brien
- Continent Chasers, a traveller and CF patient documenting travel and CF blogs, continentchasers.com
Notes[edit]
- ^ The Cystic Fibrosis Foundation recommends a diagnosis of cystic fibrosis for anyone suspected of cystic fibrosis (positive newborn screen, symptoms of cystic fibrosis, or a family history of cystic fibrosis) with sweat chloride above 60 millimoles/liter. Those with less than 30 millimoles/liter sweat chloride are unlikely to develop cystic fibrosis. For people with intermediate sweat chloride between 30 and 59 millimoles/liter, they recommend additional genetic testing.[68]
References[edit]
- ^ a b c d e f g h i j k l m n o p q r s t u O’Sullivan BP, Freedman SD (May 2009). «Cystic fibrosis». Lancet. 373 (9678): 1891–904. doi:10.1016/s0140-6736(09)60327-5. PMID 19403164. S2CID 46011502.
- ^ Allen JL, Panitch HB, Rubenstein RC (2016). Cystic Fibrosis. CRC Press. p. 92. ISBN 9781439801826. Archived from the original on 8 September 2017.
- ^ a b c d Massie J, Delatycki MB (December 2013). «Cystic fibrosis carrier screening». Paediatric Respiratory Reviews. 14 (4): 270–5. doi:10.1016/j.prrv.2012.12.002. PMID 23466339.
- ^ a b Ong T, Ramsey BW (September 2015). «Update in Cystic Fibrosis 2014». American Journal of Respiratory and Critical Care Medicine. 192 (6): 669–75. doi:10.1164/rccm.201504-0656UP. PMID 26371812.
- ^ Sencen L. «Cystic Fibrosis». NORD (National Organization for Rare Disorders). Retrieved 29 July 2022.
- ^ «Orphanet: Cystic fibrosis». www.orpha.net. Retrieved 29 July 2022.
- ^ a b c Hodson M, Geddes D, Bush A, eds. (2012). Cystic Fibrosis (3rd ed.). London: Hodder Arnold. p. 3. ISBN 978-1-4441-1369-3. Archived from the original on 8 September 2017.
- ^ Buckingham L (2012). Molecular Diagnostics: Fundamentals, Methods and Clinical Applications (2nd ed.). Philadelphia: F.A. Davis Co. p. 351. ISBN 978-0-8036-2975-2. Archived from the original on 8 September 2017.
- ^ Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D (January 2004). «Cystic fibrosis adult care: consensus conference report». Chest. 125 (1 Suppl): 1S–39S. CiteSeerX 10.1.1.562.1904. doi:10.1378/chest.125.1_suppl.1S. PMID 14734689.
- ^ a b Warnock L, Gates A (December 2015). «Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis». The Cochrane Database of Systematic Reviews. 2015 (12): CD001401. doi:10.1002/14651858.CD001401.pub3. PMC 6768986. PMID 26688006.
- ^ Nazareth D, Walshaw M (October 2013). «Coming of age in cystic fibrosis — transition from paediatric to adult care». Clinical Medicine. 13 (5): 482–6. doi:10.7861/clinmedicine.13-5-482. PMC 4953800. PMID 24115706.
- ^ a b Andersen DH (1938). «Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study». Am. J. Dis. Child. 56 (2): 344–99. doi:10.1001/archpedi.1938.01980140114013.
- ^ a b c d Egan, Schechter & Voynow 2020, «Clinical Manifestations».
- ^ a b c d e f Egan, Schechter & Voynow 2020, «Respiratory Tract».
- ^ Shteinberg M, Haq IJ, Polineni D, Davies JC (June 2021). «Cystic fibrosis». Lancet. 397 (10290): 2195–2211. doi:10.1016/S0140-6736(20)32542-3. PMID 34090606. S2CID 235327978.
- ^ Reaves J, Wallace G (2010). «Unexplained bruising: weighing the pros and cons of possible causes». Consultant for Pediatricians. 9: 201–2. Archived from the original on 22 February 2020. Retrieved 22 February 2020.
- ^ Flume PA, Mogayzel PJ, Robinson KA, Rosenblatt RL, Quittell L, Marshall BC (August 2010). «Cystic fibrosis pulmonary guidelines: pulmonary complications: hemoptysis and pneumothorax». American Journal of Respiratory and Critical Care Medicine. 182 (3): 298–306. doi:10.1164/rccm.201002-0157OC. PMID 20675678.
- ^ a b c d e f g h i j k l m n o p q r s t Mitchell RS, Kumar V, Robbins SL, et al. (2007). Robbins Basic Pathology. Saunders/Elsevier. p. 1253, 1254. ISBN 978-1-4160-2973-1.
- ^ a b c d Rowe SM, Miller S, Sorscher EJ (May 2005). «Cystic fibrosis». The New England Journal of Medicine. 352 (19): 1992–2001. doi:10.1056/NEJMra043184. PMID 15888700.
- ^ Saiman L, Siegel J (January 2004). «Infection control in cystic fibrosis». Clinical Microbiology Reviews. 17 (1): 57–71. doi:10.1128/CMR.17.1.57-71.2004. PMC 321464. PMID 14726455.
- ^ Girón RM, Domingo D, Buendía B, Antón E, Ruiz-Velasco LM, Ancochea J (October 2005). «[Nontuberculous mycobacteria in patients with cystic fibrosis]». Archivos de Bronconeumologia (in Spanish). 41 (10): 560–5. doi:10.1016/S1579-2129(06)60283-8. PMID 16266669.
- ^ Franco LP, Camargos PA, Becker HM, Guimarães RE (2009). «Nasal endoscopic evaluation of children and adolescents with cystic fibrosis». Brazilian Journal of Otorhinolaryngology. 75 (6): 806–13. doi:10.1590/S1808-86942009000600006. PMC 9446041. PMID 20209279.
- ^ Maldonado M, Martínez A, Alobid I, Mullol J (December 2004). «The antrochoanal polyp». Rhinology. 42 (4): 178–82. PMID 15626248.
- ^ Ramsey B, Richardson MA (September 1992). «Impact of sinusitis in cystic fibrosis». The Journal of Allergy and Clinical Immunology. 90 (3 Pt 2): 547–52. doi:10.1016/0091-6749(92)90183-3. PMID 1527348.
- ^ Kulczycki LL, Shwachman H (August 1958). «Studies in cystic fibrosis of the pancreas; occurrence of rectal prolapse». The New England Journal of Medicine. 259 (9): 409–12. doi:10.1056/NEJM195808282590901. PMID 13578072.
- ^ Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS (September 1998). «Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis». The New England Journal of Medicine. 339 (10): 653–8. doi:10.1056/NEJM199809033391002. PMID 9725922.
- ^ a b c Assis DN, Freedman SD (March 2016). «Gastrointestinal Disorders in Cystic Fibrosis». Clinics in Chest Medicine (Review). 37 (1): 109–118. doi:10.1016/j.ccm.2015.11.004. PMID 26857772.
- ^ Malfroot A, Dab I (November 1991). «New insights on gastro-oesophageal reflux in cystic fibrosis by longitudinal follow up». Archives of Disease in Childhood. 66 (11): 1339–1345. doi:10.1136/adc.66.11.1339. PMC 1793275. PMID 1755649.
- ^ a b c d Carroll W, Green J, Gilchrist FJ (December 2021). «Interventions for preventing distal intestinal obstruction syndrome (DIOS) in cystic fibrosis». The Cochrane Database of Systematic Reviews. 2021 (12): CD012619. doi:10.1002/14651858.CD012619.pub3. PMC 8693853. PMID 34936085.
- ^ Williams SG, Westaby D, Tanner MS, Mowat AP (October 1992). «Liver and biliary problems in cystic fibrosis». British Medical Bulletin. 48 (4): 877–92. doi:10.1093/oxfordjournals.bmb.a072583. PMID 1458306.
- ^ Colombo C, Russo MC, Zazzeron L, Romano G (July 2006). «Liver disease in cystic fibrosis». Journal of Pediatric Gastroenterology and Nutrition. 43 (Suppl 1): S49-55. doi:10.1097/01.mpg.0000226390.02355.52. PMID 16819402. S2CID 27836468.
- ^ Egan, Schechter & Voynow 2020, «Biliary Tract».
- ^ Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS, Seaquist ER (August 1994). «Insulin sensitivity in cystic fibrosis». Diabetes. 43 (8): 1020–6. doi:10.2337/diabetes.43.8.1020. PMID 8039595.
- ^ a b c de Aragão Dantas Alves C, Aguiar RA, Alves AC, Santana MA (2007). «Diabetes mellitus in patients with cystic fibrosis». Jornal Brasileiro de Pneumologia. 33 (2): 213–21. doi:10.1590/S1806-37132007000200017. PMID 17724542.
- ^ Haworth CS, Selby PL, Webb AK, Dodd ME, Musson H, McL Niven R, et al. (November 1999). «Low bone mineral density in adults with cystic fibrosis». Thorax. 54 (11): 961–7. doi:10.1136/thx.54.11.961. PMC 1745400. PMID 10525552.
- ^ McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD (October 2000). «Fertility in men with cystic fibrosis: an update on current surgical practices and outcomes». Chest. 118 (4): 1059–62. doi:10.1378/chest.118.4.1059. PMID 11035677.
- ^ Chen H, Ruan YC, Xu WM, Chen J, Chan HC (2012). «Regulation of male fertility by CFTR and implications in male infertility». Human Reproduction Update. 18 (6): 703–13. doi:10.1093/humupd/dms027. PMID 22709980.
- ^ Augarten A, Yahav Y, Kerem BS, Halle D, Laufer J, Szeinberg A, et al. (November 1994). «Congenital bilateral absence of vas deferens in the absence of cystic fibrosis». Lancet. 344 (8935): 1473–4. doi:10.1016/S0140-6736(94)90292-5. PMID 7968122. S2CID 28860665.
- ^ Gilljam M, Antoniou M, Shin J, Dupuis A, Corey M, Tullis DE (July 2000). «Pregnancy in cystic fibrosis. Fetal and maternal outcome». Chest. 118 (1): 85–91. doi:10.1378/chest.118.1.85. PMID 10893364. S2CID 32289370.
- ^ Guimbellot J, Sharma J, Rowe SM (November 2017). «Toward inclusive therapy with CFTR modulators: Progress and challenges». Pediatric Pulmonology. 52 (S48): S4–S14. doi:10.1002/ppul.23773. PMC 6208153. PMID 28881097.
- ^ Sharma J, Keeling KM, Rowe SM (August 2020). «Pharmacological approaches for targeting cystic fibrosis nonsense mutations». European Journal of Medicinal Chemistry. 200: 112436. doi:10.1016/j.ejmech.2020.112436. PMC 7384597. PMID 32512483.
- ^ Bobadilla JL, Macek M, Fine JP, Farrell PM (June 2002). «Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening». Human Mutation. 19 (6): 575–606. doi:10.1002/humu.10041. PMID 12007216. S2CID 35428054.
- ^ a b c Elborn JS (November 2016). «Cystic fibrosis». Lancet. 388 (10059): 2519–2531. doi:10.1016/S0140-6736(16)00576-6. PMID 27140670. S2CID 20948144.
- ^ Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, et al. (July 1998). «An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton». The Journal of Biological Chemistry. 273 (31): 19797–801. doi:10.1074/jbc.273.31.19797. PMID 9677412.
- ^ Travaglini KJ, Krasnow MA (August 2018). «Profile of an unknown airway cell». Nature. 560 (7718): 313–314. Bibcode:2018Natur.560..313T. doi:10.1038/d41586-018-05813-7. PMID 30097657.
- ^ a b Edwards QT, Seibert D, Macri C, Covington C, Tilghman J (November 2004). «Assessing ethnicity in preconception counseling: genetics—what nurse practitioners need to know». Journal of the American Academy of Nurse Practitioners. 16 (11): 472–80. doi:10.1111/j.1745-7599.2004.tb00426.x. PMID 15617360. S2CID 7644129.
- ^ Wang XR, Li C (May 2014). «Decoding F508del misfolding in cystic fibrosis». Biomolecules. 4 (2): 498–509. doi:10.3390/biom4020498. PMC 4101494. PMID 24970227.
- ^ Childers M, Eckel G, Himmel A, Caldwell J (2007). «A new model of cystic fibrosis pathology: lack of transport of glutathione and its thiocyanate conjugates». Medical Hypotheses. 68 (1): 101–12. doi:10.1016/j.mehy.2006.06.020. PMID 16934416.
- ^ Xu Y, Szép S, Lu Z (December 2009). «The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases». Proceedings of the National Academy of Sciences of the United States of America. 106 (48): 20515–9. Bibcode:2009PNAS..10620515X. doi:10.1073/pnas.0911412106. PMC 2777967. PMID 19918082.
- ^ Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Nauseef WM, et al. (January 2007). «A novel host defense system of airways is defective in cystic fibrosis». American Journal of Respiratory and Critical Care Medicine. 175 (2): 174–83. doi:10.1164/rccm.200607-1029OC. PMC 2720149. PMID 17082494.
- ^ Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M (January 2007). «The lactoperoxidase system links anion transport to host defense in cystic fibrosis». FEBS Letters. 581 (2): 271–8. doi:10.1016/j.febslet.2006.12.025. PMC 1851694. PMID 17204267.
- ^ Haq IJ, Gray MA, Garnett JP, Ward C, Brodlie M (March 2016). «Airway surface liquid homeostasis in cystic fibrosis: pathophysiology and therapeutic targets». Thorax. 71 (3): 284–287. doi:10.1136/thoraxjnl-2015-207588. PMID 26719229.
- ^ Verkman AS, Song Y, Thiagarajah JR (January 2003). «Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease». American Journal of Physiology. Cell Physiology. 284 (1): C2-15. doi:10.1152/ajpcell.00417.2002. PMID 12475759. S2CID 11790119.
- ^ Marieb EN, Hoehn K, Hutchinson M (2014). «22: The Respiratory System». Human Anatomy and Physiology. Pearson Education. p. 906. ISBN 978-0805361179.
- ^ a b Saiman L (2004). «Microbiology of early CF lung disease». Paediatric Respiratory Reviews. 5 (Suppl A): S367-9. doi:10.1016/S1526-0542(04)90065-6. PMID 14980298.
- ^ a b Khanolkar RA, Clark ST, Wang PW, et al. (2020). «Ecological Succession of Polymicrobial Communities in the Cystic Fibrosis Airways». mSystems. 5 (6): e00809-20. doi:10.1128/mSystems.00809-20. PMC 7716390. PMID 33262240.
- ^ a b Lorè NI, Cigana C, Riva C, De Fino I, Nonis A, Spagnuolo L, et al. (May 2016). «IL-17A impairs host tolerance during airway chronic infection by Pseudomonas aeruginosa». Scientific Reports. 6: 25937. Bibcode:2016NatSR…625937L. doi:10.1038/srep25937. PMC 4870500. PMID 27189736.
- ^ Tümmler B, Koopmann U, Grothues D, Weissbrodt H, Steinkamp G, von der Hardt H (June 1991). «Nosocomial acquisition of Pseudomonas aeruginosa by cystic fibrosis patients». Journal of Clinical Microbiology. 29 (6): 1265–7. Bibcode:1991JPoSA..29.1265A. doi:10.1002/pola.1991.080290905. PMC 271975. PMID 1907611.
- ^ Centers for Disease Control Prevention (CDC) (June 1993). «Pseudomonas cepacia at summer camps for persons with cystic fibrosis». MMWR. Morbidity and Mortality Weekly Report. 42 (23): 456–9. PMID 7684813.
- ^ Pegues DA, Carson LA, Tablan OC, FitzSimmons SC, Roman SB, Miller JM, Jarvis WR (May 1994). «Acquisition of Pseudomonas cepacia at summer camps for patients with cystic fibrosis. Summer Camp Study Group». The Journal of Pediatrics. 124 (5 Pt 1): 694–702. doi:10.1016/S0022-3476(05)81357-5. PMID 7513755.
- ^ Pankhurst CL, Philpott-Howard J (April 1996). «The environmental risk factors associated with medical and dental equipment in the transmission of Burkholderia (Pseudomonas) cepacia in cystic fibrosis patients». The Journal of Hospital Infection. 32 (4): 249–55. doi:10.1016/S0195-6701(96)90035-3. PMID 8744509.
- ^ Jones AM, Govan JR, Doherty CJ, Dodd ME, Isalska BJ, Stanbridge TN, Webb AK (June 2003). «Identification of airborne dissemination of epidemic multiresistant strains of Pseudomonas aeruginosa at a CF centre during a cross infection outbreak». Thorax. 58 (6): 525–7. doi:10.1136/thorax.58.6.525. PMC 1746694. PMID 12775867.
- ^ Høiby N (June 1995). «Isolation and treatment of cystic fibrosis patients with lung infections caused by Pseudomonas (Burkholderia) cepacia and multiresistant Pseudomonas aeruginosa». The Netherlands Journal of Medicine. 46 (6): 280–7. doi:10.1016/0300-2977(95)00020-N. PMID 7643943.
- ^ a b Pihet M, Carrere J, Cimon B, Chabasse D, Delhaes L, Symoens F, Bouchara JP (June 2009). «Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis—a review». Medical Mycology. 47 (4): 387–97. doi:10.1080/13693780802609604. PMID 19107638.
- ^ Rapaka RR, Kolls JK (2009). «Pathogenesis of allergic bronchopulmonary aspergillosis in cystic fibrosis: current understanding and future directions». Medical Mycology. 47 (Suppl 1): S331-7. doi:10.1080/13693780802266777. PMID 18668399.
- ^ «Newborn Screening for CF». Cystic Fibrosis Foundation. Retrieved 25 January 2022.
- ^ a b Egan, Schechter & Voynow 2020, «Diagnosis and Assessment».
- ^ Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, Howenstine M, McColley SA, Rock M, Rosenfeld M, Sermet-Gaudelus I, Southern KW, Marshall BC, Sosnay PR (February 2017). «Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation». J Pediatr. 181S: S4–S15.e1. doi:10.1016/j.jpeds.2016.09.064. PMID 28129811. S2CID 206410545.
- ^ Minarowski Ł, Sands D, Minarowska A, Karwowska A, Sulewska A, Gacko M, Chyczewska E (2008). «Thiocyanate concentration in saliva of cystic fibrosis patients». Folia Histochemica et Cytobiologica. 46 (2): 245–6. doi:10.2478/v10042-008-0037-0. PMID 18519245.
- ^ Stern RC (February 1997). «The diagnosis of cystic fibrosis». The New England Journal of Medicine. 336 (7): 487–91. doi:10.1056/NEJM199702133360707. PMID 9017943.
- ^ Ross LF (September 2008). «Newborn screening for cystic fibrosis: a lesson in public health disparities». The Journal of Pediatrics. 153 (3): 308–13. doi:10.1016/j.jpeds.2008.04.061. PMC 2569148. PMID 18718257.
- ^ Assael BM, Castellani C, Ocampo MB, Iansa P, Callegaro A, Valsecchi MG (September 2002). «Epidemiology and survival analysis of cystic fibrosis in an area of intense neonatal screening over 30 years». American Journal of Epidemiology. 156 (5): 397–401. doi:10.1093/aje/kwf064. PMID 12196308.
- ^ Hoch H, Sontag MK, Scarbro S, Juarez-Colunga E, McLean C, Kempe A, Sagel SD (November 2018). «Clinical outcomes in U.S. infants with cystic fibrosis from 2001 to 2012». Pediatric Pulmonology. 53 (11): 1492–1497. doi:10.1002/ppul.24165. PMID 30259702. S2CID 52845580.
- ^ Barben, Jürg, et al. (2017). «The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe». Journal of Cystic Fibrosis. 16 (2): 207–13. doi:10.1016/j.jcf.2016.12.012. PMID 28043799.
- ^ «Carrier Screening in the Age of Genomic Medicine». American College of Obstetricians and Gynecologists. 2017. Archived from the original on 25 February 2017. Retrieved 22 February 2020.
- ^ Elias S, Annas GJ, Simpson JL (April 1991). «Carrier screening for cystic fibrosis: implications for obstetric and gynecologic practice». American Journal of Obstetrics and Gynecology. 164 (4): 1077–83. doi:10.1016/0002-9378(91)90589-j. PMID 2014829.
- ^ Tabor A, Philip J, Madsen M, Bang J, Obel EB, Nørgaard-Pedersen B (June 1986). «Randomised controlled trial of genetic amniocentesis in 4606 low-risk women». Lancet. 1 (8493): 1287–93. doi:10.1016/S0140-6736(86)91218-3. PMID 2423826. S2CID 31237495.
- ^ Eddleman KA, Malone FD, Sullivan L, Dukes K, Berkowitz RL, Kharbutli Y, et al. (November 2006). «Pregnancy loss rates after midtrimester amniocentesis». Obstetrics and Gynecology. 108 (5): 1067–72. doi:10.1097/01.AOG.0000240135.13594.07. PMID 17077226. S2CID 19081825.
- ^ Davis LB, Champion SJ, Fair SO, Baker VL, Garber AM (April 2010). «A cost-benefit analysis of preimplantation genetic diagnosis for carrier couples of cystic fibrosis». Fertility and Sterility. 93 (6): 1793–804. doi:10.1016/j.fertnstert.2008.12.053. PMID 19439290.
- ^ a b «Abstracts from the 25th Italian Congress of Cystic Fibrosis and the 15th National Congress of Cystic Fibrosis Italian Society : Assago, Milan. 10 — 12 October 2019». Italian Journal of Pediatrics. 46 (Suppl 1): 32. April 2020. doi:10.1186/s13052-020-0790-z. PMC 7110616. PMID 32234058.
- ^
- ^ Davies JC, Alton EW, Bush A (December 2007). «Cystic fibrosis». BMJ. 335 (7632): 1255–9. doi:10.1136/bmj.39391.713229.AD. PMC 2137053. PMID 18079549.
- ^ Hayes D, Wilson KC, Krivchenia K, Hawkins SM, Balfour-Lynn IM, Gozal D, et al. (February 2019). «Home Oxygen Therapy for Children. An Official American Thoracic Society Clinical Practice Guideline». American Journal of Respiratory and Critical Care Medicine. 199 (3): e5–e23. doi:10.1164/rccm.201812-2276ST. PMC 6802853. PMID 30707039.
- ^ Coffey MJ, Garg M, Homaira N, Jaffe A, Ooi CY (January 2020). «Probiotics for people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 1 (1): CD012949. doi:10.1002/14651858.CD012949.pub2. PMC 6984633. PMID 31962375.
- ^ Pai VB, Nahata MC (October 2001). «Efficacy and safety of aerosolized tobramycin in cystic fibrosis». Pediatric Pulmonology. 32 (4): 314–27. doi:10.1002/ppul.1125. PMID 11568993. S2CID 30108514.
- ^ Westerman EM, Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG (March 2004). «Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study». Journal of Cystic Fibrosis. 3 (1): 23–8. doi:10.1016/j.jcf.2003.12.005. PMID 15463883.
- ^ McCoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch-Bogart GZ, Montgomery AB (November 2008). «Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis». American Journal of Respiratory and Critical Care Medicine. 178 (9): 921–8. doi:10.1164/rccm.200712-1804OC. PMC 2577727. PMID 18658109.
- ^ Ryan G, Singh M, Dwan K (March 2011). «Inhaled antibiotics for long-term therapy in cystic fibrosis». The Cochrane Database of Systematic Reviews (3): CD001021. doi:10.1002/14651858.CD001021.pub2. PMID 21412868.
- ^ «Quinsair (levofloxacin)». European Medicines Agency. Archived from the original on 26 December 2016. Retrieved 26 December 2016.
- ^ Langton Hewer SC, Smyth AR (April 2017). «Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis» (PDF). The Cochrane Database of Systematic Reviews. 4 (4): CD004197. doi:10.1002/14651858.CD004197.pub5. PMC 6478104. PMID 28440853. Archived from the original (PDF) on 24 February 2019. Retrieved 22 January 2019.
- ^ Smith S, Ratjen F, Remmington T, Waters V (May 2020). «Combination antimicrobial susceptibility testing for acute exacerbations in chronic infection of Pseudomonas aeruginosa in cystic fibrosis». The Cochrane Database of Systematic Reviews. 5 (4): CD006961. doi:10.1002/14651858.CD006961.pub5. PMC 7387858. PMID 32412092.
- ^ Hansen CR, Pressler T, Koch C, Høiby N (March 2005). «Long-term azitromycin treatment of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection; an observational cohort study». Journal of Cystic Fibrosis. 4 (1): 35–40. doi:10.1016/j.jcf.2004.09.001. PMID 15752679.
- ^ Tan KH, Mulheran M, Knox AJ, Smyth AR (March 2003). «Aminoglycoside prescribing and surveillance in cystic fibrosis». American Journal of Respiratory and Critical Care Medicine. 167 (6): 819–23. doi:10.1164/rccm.200109-012CC. PMID 12623858.
- ^ «Antibiotic resistance». Fact Sheet. World Health Organization. 31 July 2020. Retrieved 24 June 2022.
- ^ Hurley MN, Smith S, Forrester DL, Smyth AR (July 2020). «Antibiotic adjuvant therapy for pulmonary infection in cystic fibrosis». The Cochrane Database of Systematic Reviews. 7 (9): CD008037. doi:10.1002/14651858.CD008037.pub4. PMC 8407502. PMID 32671834.
- ^ Lord R, Jones AM, Horsley A (April 2020). «Antibiotic treatment for Burkholderia cepacia complex in people with cystic fibrosis experiencing a pulmonary exacerbation». The Cochrane Database of Systematic Reviews. 2020 (4): CD009529. doi:10.1002/14651858.CD009529.pub4. PMC 7117566. PMID 32239690.
- ^ Waters V, Ratjen F (June 2020). «Antibiotic treatment for nontuberculous mycobacteria lung infection in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 6 (6): CD010004. doi:10.1002/14651858.CD010004.pub5. PMC 7389742. PMID 32521055.
- ^ Kuver R, Lee SP (April 2006). «Hypertonic saline for cystic fibrosis». The New England Journal of Medicine. 354 (17): 1848–51, author reply 1848–51. doi:10.1056/NEJMc060351. PMID 16642591.
- ^ Lieberman J (July 1968). «Dornase aerosol effect on sputum viscosity in cases of cystic fibrosis». JAMA. 205 (5): 312–3. doi:10.1001/jama.205.5.312. PMID 5694947.
- ^ a b Yang C, Montgomery M, et al. (Cochrane Cystic Fibrosis and Genetic Disorders Group) (March 2021). «Dornase alfa for cystic fibrosis». The Cochrane Database of Systematic Reviews. 2021 (3): CD001127. doi:10.1002/14651858.CD001127.pub5. PMC 8094421. PMID 33735508.
- ^ Kellerman D, Rossi Mospan A, Engels J, Schaberg A, Gorden J, Smiley L (August 2008). «Denufosol: a review of studies with inhaled P2Y(2) agonists that led to Phase 3». Pulmonary Pharmacology & Therapeutics. 21 (4): 600–7. doi:10.1016/j.pupt.2007.12.003. PMID 18276176.
- ^ a b Balfour-Lynn IM, Welch K, Smith S (July 2019). «Inhaled corticosteroids for cystic fibrosis». The Cochrane Database of Systematic Reviews. 7 (4): CD001915. doi:10.1002/14651858.CD001915.pub6. PMC 6609325. PMID 31271656.
- ^ Burgess L, Southern KW (August 2014). Burgess L (ed.). «Pneumococcal vaccines for cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (8): CD008865. doi:10.1002/14651858.CD008865.pub3. PMID 25093421.
- ^ Dharmaraj P, Smyth RL (March 2014). «Vaccines for preventing influenza in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 2021 (3): CD001753. doi:10.1002/14651858.CD001753.pub3. PMC 7066935. PMID 24604671.
- ^ a b c Whiting P, Al M, Burgers L, Westwood M, Ryder S, Hoogendoorn M, et al. (March 2014). «Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis». Health Technology Assessment. 18 (18): 1–106. doi:10.3310/hta18180. PMC 4780965. PMID 24656117.
- ^ Wainwright CE (October 2014). «Ivacaftor for patients with cystic fibrosis». Expert Review of Respiratory Medicine. 8 (5): 533–8. doi:10.1586/17476348.2014.951333. PMID 25148205. S2CID 39537446.
- ^ Office of the Commissioner. «Press Announcements — FDA approves new treatment for cystic fibrosis». www.fda.gov. Archived from the original on 18 January 2017. Retrieved 16 January 2017.
- ^ «FDA approves another Vertex drug for treatment of cystic fibrosis — The Boston Globe». The Boston Globe.
- ^ «Tezacaftor (VX-661) for Cystic Fibrosis — Cystic Fibrosis News Today». Archived from the original on 29 September 2018. Retrieved 23 December 2018.
- ^ a b «Trikafta (elexacaftor, ivacaftor and tezacaftor) FDA Approval History». Drugs.com.
- ^ Office of the Commissioner (24 March 2020). «FDA approves new breakthrough therapy for cystic fibrosis». FDA. Retrieved 28 April 2022.
- ^ «FDA Accepts Vertex Application for Expansion of Trikafta to Include Children ages 6-11 | Cystic Fibrosis Foundation». www.cff.org. Retrieved 28 April 2022.
- ^ «NHS England » Landmark NHS deal to open up access to life-changing cystic fibrosis drug». www.england.nhs.uk. Retrieved 8 August 2021.
- ^ Office of the Commissioner (24 March 2020). «FDA approves new breakthrough therapy for cystic fibrosis». FDA. Retrieved 12 August 2020.
- ^ a b «Cystic Fibrosis Foundation statement on FDA approval of Trikafta™, the first triple-combination therapy for the most common CF mutation». www.cff.org. Bethesda, Md.: Cystic Fibrosis Foundation. Retrieved 12 August 2020.
- ^ a b Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. (November 2019). «Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele». The New England Journal of Medicine. 381 (19): 1809–1819. doi:10.1056/NEJMoa1908639. PMC 7282384. PMID 31697873.
- ^ Ridley K, Condren M (1 April 2020). «Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy». The Journal of Pediatric Pharmacology and Therapeutics. 25 (3): 192–197. doi:10.5863/1551-6776-25.3.192. PMC 7134581. PMID 32265602.
- ^ «Vertex prices cystic fibrosis combo treatment at $311,000-per-year». Reuters. 21 October 2019. Retrieved 23 October 2019.
- ^ Good News Network (3 November 2019). «FDA Approves the First New Cystic Fibrosis Treatment in Decades». Good News Network. Retrieved 12 August 2020.
- ^ Cheng K, Ashby D, Smyth RL (September 2017). «Ursodeoxycholic acid for cystic fibrosis-related liver disease». The Cochrane Database of Systematic Reviews. 9 (4): CD000222. doi:10.1002/14651858.CD000222.pub4. PMC 6483662. PMID 28891588.
- ^ de Vries JJ, Chang AB, Bonifant CM, Shevill E, Marchant JM (August 2018). «Vitamin A and beta (β)-carotene supplementation for cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (8): CD006751. doi:10.1002/14651858.CD006751.pub5. PMC 6513379. PMID 30091146.
- ^ Ferguson JH, Chang AB (May 2014). «Vitamin D supplementation for cystic fibrosis». The Cochrane Database of Systematic Reviews (5): CD007298. doi:10.1002/14651858.CD007298.pub4. PMID 24823922.
- ^ Okebukola PO, Kansra S, Barrett J (September 2020). «Vitamin E supplementation in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 2020 (9): CD009422. doi:10.1002/14651858.CD009422.pub4. PMC 8406985. PMID 32892350.
- ^ Jagannath VA, Thaker V, Chang AB, Price AI (June 2020). «Vitamin K supplementation for cystic fibrosis». The Cochrane Database of Systematic Reviews. John Wiley & sons, Ltd. 6 (6): CD008482. doi:10.1002/14651858.CD008482.pub6. PMC 7272115. PMID 32497260.
- ^ Watson H, Stackhouse C (April 2020). «Omega-3 fatty acid supplementation for cystic fibrosis». The Cochrane Database of Systematic Reviews. 4 (4): CD002201. doi:10.1002/14651858.CD002201.pub6. PMC 7147930. PMID 32275788.
- ^ McIlwaine M, Button B, Nevitt SJ (November 2019). «Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 2019 (11). doi:10.1002/14651858.CD003147.pub5. PMC 6953327. PMID 31774149.
- ^ Andersen JB, Qvist J, Kann T (October 1979). «Recruiting collapsed lung through collateral channels with positive end-expiratory pressure». Scandinavian Journal of Respiratory Diseases. 60 (5): 260–6. PMID 392747.
- ^ Groth S, Stafanger G, Dirksen H, Andersen JB, Falk M, Kelstrup M (July 1985). «Positive expiratory pressure (PEP-mask) physiotherapy improves ventilation and reduces volume of trapped gas in cystic fibrosis». Bulletin Européen de Physiopathologie Respiratoire. 21 (4): 339–43. PMID 3899222.
- ^ a b c Moran F, Bradley JM, Piper AJ (February 2017). «Non-invasive ventilation for cystic fibrosis». The Cochrane Database of Systematic Reviews. 2 (2): CD002769. doi:10.1002/14651858.CD002769.pub5. PMC 6464053. PMID 28218802.
- ^ Moran F, Bradley JM, Piper AJ (February 2017). «Non-invasive ventilation for cystic fibrosis». The Cochrane Database of Systematic Reviews. 2 (2): CD002769. doi:10.1002/14651858.CD002769.pub5. PMC 6464053. PMID 28218802.
- ^ «Tracheostomy Why it’s used». NHS. 3 October 2018. Retrieved 10 May 2020.
- ^ Molnar H. «Types of Tracheostomy Tubes».
- ^ Huth MM, Zink KA, Van Horn NR (2005). «The effects of massage therapy in improving outcomes for youth with cystic fibrosis: an evidence review». Pediatric Nursing. 31 (4): 328–32. PMID 16229132.
- ^ Leinwand MJ (28 December 2019). Windle ML, Odim J (eds.). «Surgical Treatment of Infections of the Lung, Pleura, and Mediastinum». Medscape. Archived from the original on 5 October 2016.
- ^ Amin R, Noone PG, Ratjen F (December 2012). «Chemical pleurodesis versus surgical intervention for persistent and recurrent pneumothoraces in cystic fibrosis». The Cochrane Database of Systematic Reviews. 12 (12): CD007481. doi:10.1002/14651858.CD007481.pub3. PMC 7208277. PMID 23235645.
- ^ Fridell JA, Vianna R, Kwo PY, Howenstine M, Sannuti A, Molleston JP, et al. (October 2005). «Simultaneous liver and pancreas transplantation in patients with cystic fibrosis». Transplantation Proceedings. 37 (8): 3567–9. doi:10.1016/j.transproceed.2005.09.091. PMID 16298663.
- ^ Belkin RA, Henig NR, Singer LG, Chaparro C, Rubenstein RC, Xie SX, et al. (March 2006). «Risk factors for death of patients with cystic fibrosis awaiting lung transplantation». American Journal of Respiratory and Critical Care Medicine. 173 (6): 659–66. doi:10.1164/rccm.200410-1369OC. PMC 2662949. PMID 16387803.
- ^ a b «Cystic Fibrosis — Pediatrics». Merck Manuals Professional Edition. Retrieved 12 August 2020.
- ^ Somaraju UR, Solis-Moya A (August 2020). «Pancreatic enzyme replacement therapy for people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (9): CD008227. doi:10.1002/14651858.CD008227.pub4. PMC 8094413. PMID 32761612.
- ^ Skolnik K, Levy RD, Wilcox PG, Quon BS (November 2016). «Coronary artery disease in cystic fibrosis: An emerging concern?». Journal of Cystic Fibrosis. 15 (6): e70–e71. doi:10.1016/j.jcf.2016.09.010. PMID 27751792.
- ^ Zirbes J, Milla CE (September 2009). «Cystic fibrosis related diabetes». Paediatric Respiratory Reviews. 10 (3): 118–23, quiz 123. doi:10.1016/j.prrv.2009.04.004. PMID 19651382.
- ^ Onady GM, Stolfi A (October 2020). «Drug treatments for managing cystic fibrosis-related diabetes». The Cochrane Database of Systematic Reviews. 2020 (10): CD004730. doi:10.1002/14651858.CD004730.pub5. PMC 8094754. PMID 33075159.
- ^ Amin R, Jahnke N, Waters V (March 2020). «Antibiotic treatment for Stenotrophomonas maltophilia in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 3 (4): CD009249. doi:10.1002/14651858.CD009249.pub5. PMC 7080526. PMID 32189337.
- ^ a b c Conwell LS, Chang AB (March 2014). «Bisphosphonates for osteoporosis in people with cystic fibrosis». The Cochrane Database of Systematic Reviews. 2014 (3): CD002010. doi:10.1002/14651858.CD002010.pub4. PMC 6718208. PMID 24627308.
- ^ Hardin DS, Rice J, Ahn C, Ferkol T, Howenstine M, Spears S, et al. (March 2005). «Growth hormone treatment enhances nutrition and growth in children with cystic fibrosis receiving enteral nutrition». The Journal of Pediatrics. 146 (3): 324–8. doi:10.1016/j.jpeds.2004.10.037. PMID 15756212.
- ^ Marks SC, Kissner DG (1997). «Management of sinusitis in adult cystic fibrosis». American Journal of Rhinology. 11 (1): 11–4. doi:10.2500/105065897781446810. PMID 9065342. S2CID 5606258.
- ^ Phillipson GT, Petrucco OM, Matthews CD (February 2000). «Congenital bilateral absence of the vas deferens, cystic fibrosis mutation analysis and intracytoplasmic sperm injection». Human Reproduction. 15 (2): 431–5. doi:10.1093/humrep/15.2.431. PMID 10655317.
- ^ Ciofu O, Lykkesfeldt J (August 2014). «Antioxidant supplementation for lung disease in cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (8): CD007020. doi:10.1002/14651858.CD007020.pub3. PMC 6777741. PMID 25102015.
- ^ a b Radtke, Thomas; Smith, Sherie; Nevitt, Sarah J.; Hebestreit, Helge; Kriemler, Susi (9 August 2022). «Physical activity and exercise training in cystic fibrosis». The Cochrane Database of Systematic Reviews. 8 (8): CD002768. doi:10.1002/14651858.CD002768.pub5. ISSN 1469-493X. PMC 9361297. PMID 35943025.
- ^ Ganesan P, Schmiedge J, Manchaiah V, Swapna S, Dhandayutham S, Kothandaraman PP (April 2018). «Ototoxicity: A Challenge in Diagnosis and Treatment». Journal of Audiology & Otology. 22 (2): 59–68. doi:10.7874/jao.2017.00360. PMC 5894487. PMID 29471610.
- ^ Green J, Gilchrist FJ, Carroll W (June 2018). «Interventions for preventing distal intestinal obstruction syndrome (DIOS) in cystic fibrosis». The Cochrane Database of Systematic Reviews. 6 (6): CD012619. doi:10.1002/14651858.cd012619. PMC 6478257. PMID 29894558.
- ^ Davis PB (March 2006). «Cystic fibrosis since 1938». American Journal of Respiratory and Critical Care Medicine. 173 (5): 475–82. doi:10.1164/rccm.200505-840OE. PMID 16126935. S2CID 1770759.
- ^ MacKenzie T, Gifford AH, Sabadosa KA, Quinton HB, Knapp EA, Goss CH, Marshall BC (August 2014). «Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation patient registry». Annals of Internal Medicine. 161 (4): 233–41. doi:10.7326/m13-0636. PMC 4687404. PMID 25133359.
- ^ «Canadian Cystic Fibrosis Patient Data Registry Report» (PDF). Canadian Cystic Fibrosis Foundation. 2007. Archived from the original (PDF) on 15 July 2010. Retrieved 14 March 2010.
- ^ «Annual Data Report 2016 Cystic Fibrosis Foundation Patient Registry» (PDF). p. 4. Archived from the original (PDF) on 19 June 2018. Retrieved 19 June 2018.
- ^ «Cystic Fibrosis Patient Registry Annual Data Report 2009» (PDF). Cystic Fibrosis Foundation. 2009. Archived from the original (PDF) on 5 January 2012.
- ^ Yu H, Nasr SZ, Deretic V (April 2000). «Innate lung defenses and compromised Pseudomonas aeruginosa clearance in the malnourished mouse model of respiratory infections in cystic fibrosis». Infection and Immunity. 68 (4): 2142–7. doi:10.1128/IAI.68.4.2142-2147.2000. PMC 97396. PMID 10722612.
- ^ Ratjen F, Döring G (February 2003). «Cystic fibrosis». Lancet. 361 (9358): 681–9. doi:10.1016/S0140-6736(03)12567-6. PMID 12606185. S2CID 24879334.
- ^ Rosenstein BJ, Zeitlin PL (January 1998). «Cystic fibrosis». Lancet. 351 (9098): 277–82. doi:10.1016/S0140-6736(97)09174-5. PMID 9457113. S2CID 44627706.
- ^ Schmitz TG, Goldbeck L (February 2006). «The effect of inpatient rehabilitation programmes on quality of life in patients with cystic fibrosis: a multi-center study». Health and Quality of Life Outcomes. 4: 8. doi:10.1186/1477-7525-4-8. PMC 1373610. PMID 16457728.
- ^ Hegarty M, Macdonald J, Watter P, Wilson C (July 2009). «Quality of life in young people with cystic fibrosis: effects of hospitalization, age and gender, and differences in parent/child perceptions». Child. 35 (4): 462–8. doi:10.1111/j.1365-2214.2008.00900.x. PMID 18991968.
Havermans T, Vreys M, Proesmans M, De Boeck C (January 2006). «Assessment of agreement between parents and children on health-related quality of life in children with cystic fibrosis». Child. 32 (1): 1–7. doi:10.1111/j.1365-2214.2006.00564.x. PMID 16398786. - ^ Moorcroft AJ, Dodd ME, Webb AK (1998). «Exercise limitations and training for patients with cystic fibrosis». Disability and Rehabilitation. 20 (6–7): 247–53. doi:10.3109/09638289809166735. PMID 9637933.
- ^ «Medications». Cystic Fibrosis Canada. 2011. No. 10684-5100 RR0001. Archived from the original on 4 September 2011.
- ^ Araújo FG, Novaes FC, Santos NP, Martins VC, Souza SM, Santos SE, Ribeiro-dos-Santos AK (January 2005). «Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the North of Brazil». Brazilian Journal of Medical and Biological Research. 38 (1): 11–5. doi:10.1590/S0100-879X2005000100003. PMID 15665983.
- ^ Tobias E (2011). Essential Medical Genetics. John Wiley & Sons. p. 312. ISBN 978-1-118-29370-6. Archived from the original on 17 April 2016.
- ^ «The Canadian Facts & Figures on Cystic Fibrosis». cysticfibrosis.ca. Archived from the original on 16 June 2013.
- ^ «Genetic Carrier Testing». Cystic Fibrosis Foundation. 2007. Archived from the original on 23 March 2010.
- ^ Rosenstein BJ, Cutting GR (April 1998). «The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel». The Journal of Pediatrics. 132 (4): 589–95. doi:10.1016/S0022-3476(98)70344-0. PMID 9580754.
- ^ Hamosh A, FitzSimmons SC, Macek M, Knowles MR, Rosenstein BJ, Cutting GR (February 1998). «Comparison of the clinical manifestations of cystic fibrosis in black and white patients». The Journal of Pediatrics. 132 (2): 255–9. doi:10.1016/S0022-3476(98)70441-X. PMID 9506637.
- ^ Farrell P, Joffe S, Foley L, Canny GJ, Mayne P, Rosenberg M (September 2007). «Diagnosis of cystic fibrosis in the Republic of Ireland: epidemiology and costs». Irish Medical Journal. 100 (8): 557–60. PMID 17955689. Archived from the original on 3 December 2013.
- ^ Hytönen M, Patjas M, Vento SI, Kauppi P, Malmberg H, Ylikoski J, Kere J (December 2001). «Cystic fibrosis gene mutations deltaF508 and 394delTT in patients with chronic sinusitis in Finland». Acta Oto-Laryngologica. 121 (8): 945–7. doi:10.1080/000164801317166835. PMID 11813900.
- ^ «WHO | Genes and human disease». Who.int. 7 December 2010. Archived from the original on 20 October 2012. Retrieved 23 January 2013.
- ^ Russell P (2011). Biology: the dynamic science (2nd ed.). Belmont, CA: Brooks/Cole, Cengage Learning. p. 304. ISBN 978-0-538-49372-7. Archived from the original on 17 April 2016.
- ^ «Genetic testing for cystic fibrosis Genetic Testing for Cystic Fibrosis». Consensus Development Conference Statement. National Institutes of Health. 14–16 April 1997. Archived from the original on 27 March 2009.
- ^ Rosenfeld M, Davis R, FitzSimmons S, Pepe M, Ramsey B (May 1997). «Gender gap in cystic fibrosis mortality». American Journal of Epidemiology. 145 (9): 794–803. doi:10.1093/oxfordjournals.aje.a009172. PMID 9143209.
- ^ Coakley RD, Sun H, Clunes LA, Rasmussen JE, Stackhouse JR, Okada SF, et al. (December 2008). «17beta-Estradiol inhibits Ca2+-dependent homeostasis of airway surface liquid volume in human cystic fibrosis airway epithelia». The Journal of Clinical Investigation. 118 (12): 4025–35. doi:10.1172/JCI33893. PMC 2582929. PMID 19033671.
- ^ Verma N, Bush A, Buchdahl R (October 2005). «Is there still a gender gap in cystic fibrosis?». Chest. 128 (4): 2824–34. doi:10.1378/chest.128.4.2824. PMID 16236961.
- ^ Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W (September 2009). «Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality». Diabetes Care. 32 (9): 1626–31. doi:10.2337/dc09-0586. PMC 2732133. PMID 19542209.
- ^ «CF worse for women ‘due to effect of estrogen’«. The Irish Times. 8 August 2010. Archived from the original on 11 August 2010.
- ^ Wennberg C, Kucinskas V (1994). «Low frequency of the delta F508 mutation in Finno-Ugrian and Baltic populations». Human Heredity. 44 (3): 169–71. doi:10.1159/000154210. PMID 8039801.
- ^ Kere J, Savilahti E, Norio R, Estivill X, de la Chapelle A (September 1990). «Cystic fibrosis mutation delta F508 in Finland: other mutations predominate». Human Genetics. 85 (4): 413–5. doi:10.1007/BF02428286. PMID 2210753. S2CID 38364780.
- ^ Wiuf C (August 2001). «Do delta F508 heterozygotes have a selective advantage?». Genetical Research. 78 (1): 41–7. CiteSeerX 10.1.1.174.7283. doi:10.1017/S0016672301005195. PMID 11556136.
- ^ Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ (October 1994). «Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model». Science. 266 (5182): 107–9. Bibcode:1994Sci…266..107G. doi:10.1126/science.7524148. PMID 7524148.
- ^ Alfonso-Sánchez MA, Pérez-Miranda AM, García-Obregón S, Peña JA (June 2010). «An evolutionary approach to the high frequency of the Delta F508 CFTR mutation in European populations». Medical Hypotheses. 74 (6): 989–92. doi:10.1016/j.mehy.2009.12.018. PMID 20110149.
- ^ Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, Evans MJ (January 1995). «The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study». The Journal of Physiology. 482 (Pt 2): 449–54. doi:10.1113/jphysiol.1995.sp020531. PMC 1157742. PMID 7714835.
- ^ Högenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, et al. (December 2000). «Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion». American Journal of Human Genetics. 67 (6): 1422–7. doi:10.1086/316911. PMC 1287919. PMID 11055897.
- ^ Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, et al. (May 1998). «Salmonella typhi uses CFTR to enter intestinal epithelial cells». Nature. 393 (6680): 79–82. Bibcode:1998Natur.393…79P. doi:10.1038/30006. PMID 9590693. S2CID 5894247.
- ^ Modiano G, Ciminelli BM, Pignatti PF (March 2007). «Cystic fibrosis and lactase persistence: a possible correlation». European Journal of Human Genetics. 15 (3): 255–9. doi:10.1038/sj.ejhg.5201749. hdl:2108/29185. PMID 17180122. S2CID 4650571.
- ^ Poolman EM, Galvani AP (February 2007). «Evaluating candidate agents of selective pressure for cystic fibrosis». Journal of the Royal Society, Interface. 4 (12): 91–8. doi:10.1098/rsif.2006.0154. PMC 2358959. PMID 17015291.
- ^ Williams N (2006). «Footprint fears for new TB threat». Current Biology. 16 (19): R821–R822. doi:10.1016/j.cub.2006.09.009. S2CID 2346727.
- ^ Tobacman JK (June 2003). «Does deficiency of arylsulfatase B have a role in cystic fibrosis?». Chest. 123 (6): 2130–9. doi:10.1378/chest.123.6.2130. PMID 12796199.
- ^ a b c Busch R (1990). «On the History of Cystic Fibrosis». Acta Universitatis Carolinae. Medica. 36 (1–4): 13–15. PMID 2130674.
- ^ Fanconi G, Uehlinger E, Knauer C (1936). «Das coeliakiesyndrom bei angeborener zysticher pankreasfibromatose und bronchiektasien». Wien. Med. Wochenschr. 86: 753–756.
- ^ Di Sant’Agnese PA, Darling RC, Perera GA, Shea E (November 1953). «Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease». Pediatrics. 12 (5): 549–563. doi:10.1542/peds.12.5.549. PMID 13111855. S2CID 42514224.
- ^ Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. (September 1989). «Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA». Science. 245 (4922): 1066–1073. Bibcode:1989Sci…245.1066R. doi:10.1126/science.2475911. PMID 2475911. S2CID 84566748.
- ^ Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. (September 1989). «Identification of the cystic fibrosis gene: chromosome walking and jumping». Science. 245 (4922): 1059–1065. Bibcode:1989Sci…245.1059R. doi:10.1126/science.2772657. PMID 2772657.
- ^ Freudenheim M (22 December 2009). «Tool in Cystic Fibrosis Fight: A Registry». The New York Times. pp. D1. Archived from the original on 24 May 2013. Retrieved 21 December 2009.
- ^ Lee TW, Southern KW, Perry LA, Penny-Dimri JC, Aslam AA (June 2016). Southern KW (ed.). «Topical cystic fibrosis transmembrane conductance regulator gene replacement for cystic fibrosis-related lung disease». The Cochrane Database of Systematic Reviews. 2016 (6): CD005599. doi:10.1002/14651858.CD005599.pub5. PMC 8682957. PMID 27314455.
- ^ Alton EW, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. (September 2015). «Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial». The Lancet. Respiratory Medicine. 3 (9): 684–691. doi:10.1016/S2213-2600(15)00245-3. PMC 4673100. PMID 26149841.
- ^ Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD (November 2002). «Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis». American Journal of Respiratory Cell and Molecular Biology. 27 (5): 619–27. doi:10.1165/rcmb.2001-0004oc. PMID 12397022. S2CID 8714332.
- ^ Tate S, Elborn S (March 2005). «Progress towards gene therapy for cystic fibrosis». Expert Opinion on Drug Delivery. 2 (2): 269–80. doi:10.1517/17425247.2.2.269. PMID 16296753. S2CID 30948229.
- ^ Online Mendelian Inheritance in Man (OMIM): CYSTIC FIBROSIS; CF — 219700
- ^ Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, et al. (December 2013). «Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients». Cell Stem Cell. 13 (6): 653–8. doi:10.1016/j.stem.2013.11.002. PMID 24315439.
- ^ Hraiech S, Brégeon F, Rolain JM (2015). «Bacteriophage-based therapy in cystic fibrosis-associated Pseudomonas aeruginosa infections: rationale and current status». Drug Design, Development and Therapy. 9: 3653–63. doi:10.2147/DDDT.S53123. PMC 4509528. PMID 26213462.
- ^ Trend S, Fonceca AM, Ditcham WG, Kicic A, Cf A (November 2017). «The potential of phage therapy in cystic fibrosis: Essential human-bacterial-phage interactions and delivery considerations for use in Pseudomonas aeruginosa-infected airways». Journal of Cystic Fibrosis. 16 (6): 663–670. doi:10.1016/j.jcf.2017.06.012. PMID 28720345.
- ^ a b Ramsey BW, Downey GP, Goss CH (May 2019). «Update in Cystic Fibrosis 2018». American Journal of Respiratory and Critical Care Medicine. 199 (10): 1188–1194. doi:10.1164/rccm.201902-0310UP. PMC 6519861. PMID 30917288. ProQuest 2230820891.
- ^ Dietz HC (August 2010). «New therapeutic approaches to mendelian disorders». The New England Journal of Medicine. 363 (9): 852–63. doi:10.1056/NEJMra0907180. PMID 20818846. S2CID 5809127. Free full text
- ^ a b Office of the Commissioner (24 October 2019). «FDA approves new breakthrough therapy for cystic fibrosis». FDA. Retrieved 13 November 2019.
- ^ «CFTR Modulator Therapies». Bethesda, Md.: Cystic Fibrosis Foundation. Retrieved 13 November 2019.
- ^ Sherrard LJ, McGrath SJ, McIlreavey L, Hatch J, Wolfgang MC, Muhlebach MS, Gilpin DF, Elborn JS, Tunney MM (February 2016). «Production of extended-spectrum beta-lactamases and the potential indirect pathogenic role of Prevotella isolates from the cystic fibrosis respiratory microbiota». Int J Antimicrob Agents. 47 (2): 140–145. doi:10.1016/j.ijantimicag.2015.12.004. PMC 4746055. PMID 26774156.
Further reading[edit]
- Egan ME, Schechter MS, Voynow JA (2020). «Cystic Fibrosis». In Kliegman RM, St Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM (eds.). Nelson Textbook of Pediatrics. Elsevier. pp. 2282–2297. ISBN 978-0-323-56890-6.
External links[edit]
- Search GeneCards for genes involved in cystic fibrosis
- Cystic Fibrosis Mutation Database
- «Cystic Fibrosis». MedlinePlus. U.S. National Library of Medicine.
наследственное заболевание, характеризующееся системным поражением экзокринных желез и проявляющееся тяжелыми расстройствами функций органов дыхания, желудочно-кишечного тракта и ряда других органов и систем. Частота муковисцидоза в СССР, по данным разных авторов, составляет от 1:2500 до 1:8000 новорожденных, в других странах мира от 1:2000 до 1:90000. Наиболее часто М. встречается у жителей Северной и Центральной Европы.
Этиология и патогенез. Заболевание наследуется по аутосомно-рецессивному типу. В патогенезе М. выделяют три основных звена: поражение экзокринных желез, нарушение электролитного обмена, поражение соединительной ткани. При М экзокринные железы продуцируют секрет, характеризующийся повышенной вязкостью, высоким содержанием белка и некоторых электролитов и обладающий пониженными ферментными свойствами. Вязкий секрет закупоривает протоки желез, что приводит к их расширению и образованию кист, которые в последующем могут инфицироваться.
Электролитные нарушения проявляются высоким содержанием кальция в секрете экзокринных желез, накоплением натрия и хлора в ногтях, волосах, слезной жидкости, высоким содержанием натрия и хлора в поте.
Фибробласты соединительной ткани накапливают Гликозаминогликаны и продуцируют цилиарный фактор (фактор Спока), нарушающий движение ресничек эпителия слизистой оболочки трахеи и бронхов. Поражение фибробластов, являющихся активным элементом соединительной ткани, играющих важную роль в синтезе гликозаминогликанов, в росте и дифференцировке коллагеновых волокон, организации волокнистых структур, приводит к раннему развитию склеротических процессов в органах. В случае присоединения вторичной инфекции и развития воспаления склероз прогрессирует значительно быстрее.
Патологическая анатомия. Морфологические изменения в различных органах однотипны: склероз, кисты, участки воспаления. Так, в поджелудочной железе выявляют диффузный фиброз, кисты мелких и средних протоков, воспалительную инфильтрацию (рис. 1). Островки Лангерганса увеличены в размерах. С возрастом склероз прогрессирует, появляются участки липоматоза, кисты уменьшаются в размерах. Железы слизистой оболочки кишечника кистозно расширены, например бокаловидные клетки принимают шаровидную форму из-за скопления в них секрета, отмечается фиброз окружающей ткани.
В печени выявляются поля развития соединительной ткани между дольками и под капсулой, кистозно расширенные мелкие желчные протоки, признаки жировой и белковой дистрофии. В ряде случаев обнаруживаются гипоплазия желчного пузыря, атрезия пузырного протока или их сочетание.
В легких выражен пневмосклероз, имеются участки эмфиземы, зоны ателектаза, воспалительные изменения. Бронхи и бронхиолы заполнены слизисто-гнойными массами, бронхиальные железы увеличены в размерах за счет слизистого содержимого, выпячиваются в просвет бронхов. При длительном течении процесса слизисто-гнойные пробки полностью закрывают просветы бронхов, в стенках бронхов нарастают атрофические изменения с последующим развитием бронхоэктазов. Отмечается перибронхиальный и периваскулярный склероз. На фоне таких изменений нередко возникает стафилококковая деструктивная пневмония.
Клиническая картина многообразна и зависит от преимущественного поражения тех или иных экзокринных желез, степени склероза в различных органах, наличия осложнений и возраста больных. Выделяют легочную, кишечную, смешанную (кишечно-легочную), атипичные формы М. и мекониальную непроходимость. Деление на клинические формы условно, т.к. при чисто легочной форме отмечается снижение ферментативной активности желез желудочно-кишечного тракта, а при чисто кишечной постепенно формируется пневмосклероз и с возрастом присоединяется выраженный легочный синдром. Мекониальная непроходимость — первый признак тяжелой кишечной формы М. у новорожденного первых дней жизни; в последующем развивается смешанная форма. К атипичным относят формы, протекающие с изолированным поражением отдельных экзокринных желез (например, печени), и легкие стертые формы. Ведущим синдромом, определяющим тяжесть и прогноз заболевания, является легочный.
Первые симптомы заболевания возникают чаще на первом году жизни, реже в более старшем возрасте. Чем раньше проявляется заболевание, тем тяжелее оно протекает и тем серьезнее прогноз.
Легочный синдром при М. начинается обычно с пневмонии (Пневмония). Наиболее частыми возбудителями ее у детей, больных М., являются стафилококк и синегнойная палочка. Иногда до появления симптомов пневмонии у ребенка в течение длительного времени отмечается сухой приступообразный кашель. При развитии пневмонии кашель становится мучительным, приступообразным, коклюшеподобным. Кашель обусловлен раздражением слизистой оболочки бронхов вязкой, с трудом отделяющейся мокротой. Слюна, слизь в носу у больных тоже вязкая и липкая.
Вязкая мокрота вызывает сужение бронхов, что ведет к эмфиземе (рис. 2), или полностью закупоривает бронхи, что обусловливает развитие ателектаза легкого (Ателектаз лёгкого). Ателектазы длительно не расправляются, нередко бывают множественными или «мигрирующими» (расправляются в одних сегментах легких и возникают в других). Чаще ателектазы наблюдаются у детей раннего возраста. Отмечается укорочение перкуторного звука в паравертебральных областях, над пневмоническими фокусами и ателектазами. Определяются участки с коробочным оттенком перкуторного звука. С обеих сторон выслушивается множество сухих и разнокалиберных влажных хрипов; в случае скопления большого количества мокроты в бронхах хрипы в отдельных участках легких могут не выслушиваться. Рано развивается Пневмосклероз.
Пневмонические очаги, ателектазы, участки эмфиземы, пневмосклероза создают типичную для М. пеструю клиническую и рентгенологическую картину. Процесс в легких всегда двусторонний. Характерно затяжное рецидивирующее течение пневмонии, нередко с абсцедированием и ранним появлением бронхоэктазов. У некоторых больных развивается деструктивная пневмония с вовлечением в процесс плевры.
При тяжелом течении легочного синдрома процесс неуклонно прогрессирует, часто возникают обострения. Мокрота быстро становится гнойной. Наблюдаются деформации грудной клетки: бочкообразная, килевидная, редко воронкообразная. Дыхание жесткое, иногда амфорическое (над полостями). Выслушиваются сухие и влажные хрипы, при обструктивном синдроме — сухие свистящие хрипы. При скоплении мокроты, чаще в нижних отделах легких, дыхание ослаблено, хрипов мало.
Постепенно нарастают симптомы дыхательной недостаточности (Дыхательная недостаточность): одышка в покое, цианоз, тахикардия. Развиваются симптомы легочного сердца (Лёгочное сердце), легочной и сердечной недостаточности. Появляется деформация пальцев рук и ног в виде барабанных палочек, ногтей в виде часовых стекол. Дети отстают в физическом развитии, появляются дистрофические изменения кожи (рис. 3), она становится сухой, шелушится, истончается, волосы делаются ломкими, теряют блеск, истончаются, приобретают сероватый оттенок, ногти утолщаются, легко ломаются, слоятся, на них появляются линейные утолщения в виде полосочек, беловатых пятен.
При рентгенологическом исследовании выявляют эмфизему легких, диффузный пневмосклероз с фиброзными уплотнениями при корневых зонах (рис. 4). На бронхограммах определяют деформацию бронхов, их «обрубленность», сближение, цилиндрические, мешотчатые и каплевидные бронхоэктазы (рис. 5). При бронхоскопии устанавливают диффузный катарально-гнойный эндобронхит. Поражение бронхов всегда двустороннее, диффузное. У некоторых больных обнаруживают пороки развития бронхов (Бронхи) в виде дополнительного бронха, бронхомегалии, отхождение бронха средней доли правого легкого от верхнедолевого бронха. Высокая частота пороков развития при М. свидетельствует об их связи с основным заболеванием.
У детей с легочным синдромом значительно нарушены функция внешнего дыхания по обструктивно-рестриктивному типу, рано развивается дыхательный, метаболический или смешанный Ацидоз. При более легком течении легочного синдрома процесс протекает по типу медленно прогрессирующего Бронхита с редкими обострениями, физическое развитие детей удовлетворительное.
Кишечный синдром обусловлен резким снижением активности ферментов поджелудочной железы и кишечника. Характерным для него является нарушение расщепления и всасывания жиров, белков, углеводов пищи. У больных учащается стул до 2—5 раз в сутки, фекалии становятся обильными и жирными, имеют серый цвет, резкий зловонный запах. Иногда кал бывает плотным, скудным («овечий» кал), появляется склонность к запорам (возможно выпадение прямой кишки), что вызвано повышенной вязкостью секрета поджелудочной железы и кишечника и хорошим всасыванием воды в кишечнике. Однако цвет фекалий остается светло-серым, в конце дефекации выделяется капля жира. Дети теряют в массе, несмотря на сохраненный аппетит. При копрологическом исследовании обнаруживают в большом количестве нейтральный жир, немного жирных кислот и мыла, нерасщепленные мышечные волокна и крахмал.
Мекониальная непроходимость развивается в первые сутки жизни ребенка и обусловлена образованием в тонкой кишке плотных вязких мекониевых пробок, имеющих иногда протяженность до 3—8 см. Если пробка не размывается, то возникают некроз участка тонкой кишки и перитонит, от которого ребенок, как правило, погибает.
Легкие стертые формы М. протекают по типу хронического бронхита, хронического Панкреатита с ферментативной недостаточностью, хронического энтероколита (см. Энтерит). В некоторых случаях на фоне склероза поджелудочной железы развивается Диабет сахарный.
У многих больных М. выявляют увеличение размеров печени, жировую, белковую или белково-жировую дистрофию гепатоцитов (по данным аутопсии, биопсии и эхографии печени). Эти изменения длительно существуют, не проявляясь клинически, при этом уровни ферментов печени также остаются нормальными. В ряде случаев поражение печени может привести к циррозу, для которого характерно медленное прогрессирующее развитие синдрома портальной гипертензии (см. Портальная гипертензия) при длительно сохраняющейся нормальной функции гепатоцитов. Значительно реже развивается билиарный цирроз печени.
Нередко при М. наблюдаются обменные нефропатии, что проявляется гиперфосфатурией, оксалурией, протеинурией, цистинурией: на фоне этих изменений может развиться пиелонефрит. У больных М. отмечается дисфункция коры надпочечников, характерна задержка полового развития.
Муковисцидоз может осложниться плевритом, пневмотораксом, деструкцией легких, кровохарканьем, кишечной непроходимостью, выпадением прямой кишки, сахарным диабетом, хроническим легочным сердцем, вторичной дисахаридазной недостаточностью, синдромом целиакии.
Диагноз ставят на основании анамнеза (отягощенная наследственность, раннее начало заболевания), клинических симптомов и результатов лабораторных исследований. Достоверным диагноз М. считают при обнаружении высокого содержания натрия и хлора в поте, высокой концентрации натрия в ногтевых пластинках пальцев рук. Патогномоничным для М. является содержание натрия и хлора в поте у детей до 1 года свыше 40 ммоль/л, у детей старше одного года и у взрослых — свыше 60 ммоль/л; концентрация натрия в ногтевых пластинках рук свыше 80 ммоль/кг (для любого возраста).
При исследовании содержания натрия и хлора в поте для усиления потоотделения проводят электрофорез с пилокарпином участка кожи на предплечье, поэтому пробу нередко называют пилокарпиновой. Кожу внутренней поверхности предплечья очищают спиртом и дистиллированной водой, затем на этот участок накладывают два электрода: под отрицательный электрод кладут марлевую прокладку, смоченную раствором, который содержит хлорид натрия, под положительный электрод (анод) — марлевую прокладку, смоченную 0,5% раствором пилокарпина. Электрофорез продолжается 10 мин при силе тока 4 мА (силу тока постепенно увеличивают от 0 до 4 мА), после чего силу тока постепенно снижают и электроды снимают. Участок кожи, на котором был электрод с пилокарпиновой прокладкой, обрабатывают дистиллированной водой, высушивают, после чего на него накладывают кусочек предварительно взвешенной беззольной фильтровальной бумаги (3×4 см). Для предупреждения испарения пота фильтровальную бумагу накрывают полиэтиленовой пленкой, которую по краям приклеивают лейкопластырем. Пот собирают в течение 30 мин, затем фильтровальную бумагу снимают пинцетом и тут же взвешивают для определения количества впитавшегося в нее пота. Для получения достоверного результата количество пота должно быть не менее 100 мг.
После взвешивания фильтровальную бумагу в бюксе заливают 10 мл дистиллированной воды и оставляют для элюирования электролитов не менее чем на 4 ч. Можно элюировать и до 20 ч, чтобы дальнейшие исследования проводить на следующий день, но надо удостовериться, что крышка в бюксе тщательно притерта и вода не испаряется. Концентрацию натрия в элюате определяют на пламенном фотометре, а концентрацию хлора — титрованием по методу Мора.
Для определения натрия в ногтевых пластинках используют методику Копито и Швахмана в модификации Вельтищева и Глотовой. Для исследования следует использовать срезанные кусочки ногтей с пальцев рук (у детей в возрасте до 1 года можно с рук и ног). Их очищают, моют дистиллированной водой, затем высушивают в термостате 1 ч при 37°. К навеске ногтей (10 мг) добавляют 0,2 мл концентрированной азотной килоты, после чего сосуд ставят на водяную баню до полного растворения ногтей (2—3 мин). Добавляют 4,8 мл дистиллированной воды, тщательно смешивают, затем на пламенном фотометре определяют концентрацию натрия и пересчитывают в ммоль/кг ногтей. Такое исследование удобно тем, что может быть проведено в другом медицинском учреждении, где имеется пламенный фотометр, и незаменимо в тех случаях, когда трудно получить для исследования пот (недоношенные дети, дети с экземой и другими заболеваниями кожи).
В сомнительных случаях, при наличии клинических признаков М. и близких к норме концентрациях натрия и хлора в поте, можно использовать нагрузочную пробу с дезоксикортикостерона ацетатом (ДОКСА). Для этого после определения концентрации натрия и хлора в поте (пилокар-пиновая проба) назначают гипохлоридную диету на 4 дня (у детей до 2 лет диеты не требуется); на 4-й день в 20 ч и на 5-й день в 8 ч внутримышечно вводят 0,5% масляный раствор ДОКСА (детям до 6 мес. 0,2 мл, от 6 мес. до 3 лет 0,4 мл, от 3 до 7 лет 0,6 мл, от 7 до 10—12 лет 0,8 мл, старше 12 лет и взрослым 1,0 мл). Через 4—6 ч после второй инъекций ДОКСА собирают пот для повторного исследования. У детей, не страдающих М., концентрация натрия и хлора в поте после нагрузки с ДОКСА снижается более чем на 25% по сравнению с исходной, у детей, больных М., остается без изменений, незначительно снижается или даже повышается.
Дифференциальный диагноз легочной формы М. проводят с хронической пневмонией и наследственно обусловленными поражениями легких (идиопатическим гемосидерозом легких, синдромом Картагенера, хронической пневмонией при иммунодефицитных состояниях, болезнью Марфана). Кишечную форму муковисцидоза дифференцируют с другими синдромами нарушенного кишечного всасывания — Глютеновая болезнь (целиакия), Дисахаридазная недостаточность, Энтеропатия экссудативная, дисбактериоз кишечника. Основным при проведении дифференциального диагноза является исследование электролитов.
Лечение. Диета больного М. должна соответствовать возрасту, содержать повышенное на 10—15% количество белка и нормальное количество жиров и углеводов. При этом в рацион включают только легко усваиваемые жиры (сливочное и растительное масло). Пища не должна содержать грубую клетчатку. У детей с вторичной лактазной недостаточностью исключают молоко. При выраженном кишечном синдроме и симптомах, обусловленных недостаточностью различных витаминов, парентерально назначают витамины.
При кишечном синдроме с заместительной целью применяют ферментные препараты: панкреатин, панзинорм, котазим-форте, фестал и др. Доза фермента зависит от тяжести заболевания и подбирается индивидуально. Критериями достаточности дозы являются исчезновение болей в животе, нормализация стула, отсутствие нейтрального жира при копрологическом исследовании. Ферментные препараты принимают во время еды.
Лечение легочного синдрома включает мероприятия по уменьшению вязкости мокроты и улучшению дренажа бронхов, антибактериальную терапию, борьбу с интоксикацией и гипоксией, гиповитаминозом, сердечной недостаточностью. Для уменьшения вязкости мокроты применяют ингаляции ферментных препаратов (химопсина, химотрипсина, кристаллического фибринолизина) или муколитических препаратов, ацетилцистеина, мукосольвина. С целью разжижения мокроты ацетилцистеин и мукосольвин можно назначать внутримышечно, а мукосольвин также и внутрь. Более слабый разжижающий эффект оказывают бромгексин и мукалтин. Для улучшения дренажа бронхов проводят вибрационный массаж грудной клетки, лечебную гимнастику, постуральный дренаж, v детей раннего возраста мокроту удаляют электроотсосом.
При обострении легочного процесса назначают антибактериальную терапию сроком не менее чем на 3—4 нед. Антибиотики подбирают с учетом антибиотикограммы, но если это исследование провести невозможно, то основываются на том, что наиболее часто возбудителями воспалительного процесса в легких у больных М. являются стафилококк и синегнойная палочка. Одновременно с антибиотиками используют противогрибковые (нистатин, чеворин) и антигистаминные препараты. В период обострения проводят также УВЧ-, СВЧ-герапию. а затем электрофорез препаратов пода и магния (препараты кальция противопоказаны, т.к. они усиливают пневмосклероз); назначают поливитамины, для уменьшения легочной гипертензии — эуфиллин по 7—10 мг/кг массы тела в сутки внутрь (дозу делят на 3 приема) в течение 4—5 нед. от начала обострения. Показаны препараты, улучшающие метаболизм миокарда: оротат калия, кокарбоксилаза. При декомпенсации легочного сердца применяют сердечные гликозиды (дигоксин), глюкокортикоиды (по 1—1,5 мг/кг в сутки в пересчете на преднизолон с учетом суточного ритма надпочечников в течение 3—4 нед.). Глюкокортикоиды в такой же дозе назначают и при быстром прогрессировании пневмосклероза, а при хронической надпочечниковой недостаточности, развившейся в результате гнойной интоксикации и гипоксии, — в дозе 0,4 мг/кг в сутки по преднизолону с учетом суточного ритма.
Дети, больные М., находятся под диспансерным наблюдением участкового врача и пульмонолога, т. к., несмотря на генерализацию процесса и изменения во многих органах и системах, поражение органов дыхания отмечается у большинства больных и именно оно определяет тяжесть и прогноз заболевания. Родители должны быть обучены уходу за больным и таким методам лечения, как массаж, лечебная гимнастика, аэрозольтерапия. Задачами диспансерного наблюдения являются контроль за функциональным состоянием бронхолегочной, сердечно-сосудистой систем, желудочно-кишечного тракта, почек, печени и правильностью дозы ферментных препаратов, своевременное лечение при обострении заболевания, проведение общеукрепляющей терапии, в периоде ремиссии — санация очагов хронической инфекции. Лечение осуществляют амбулаторно и в домашних условиях, где ребенку может быть обеспечен индивидуальный уход и исключена реинфекция. Лишь при тяжелом состоянии или наличии осложнений показана госпитализация. Интенсивную терапию проводят больным с дыхательной недостаточностью II—III степени, с декомпенсацией легочного сердца, с плевральными осложнениями, деструкцией легких, кровохарканьем Оперативное вмешательство показано при мекониальной непроходимости, при отсутствии эффекта от консервативной терапии при кишечной непроходимости у детей более старшего возраста, иногда при деструкции в легких. При бронхоэктазах хирургическое лечение не рекомендуется, т.к. процесс всегда распространенный.
Больные муковисцидозом дети получают все лекарства для амбулаторного лечения бесплатно.
Детям с легкими и среднетяжелыми кишечными формами М. показано санаторное лечение. Для детей с легочной формой М. полезно санаторное лечение, если есть возможность создать специальные группы. Больных М. рекомендуется направлять в местные санатории. Критериями отбора в санаторий являются компенсация кишечных расстройств при назначении ферментных препаратов, отсутствие декомпенсации легочного сердца и воспалительного процесса в легких.
Пребывание больных М. в детских дошкольных учреждениях нецелесообразно. Посещение школы при хорошем и удовлетворительном состоянии возможно, но необходим дополнительный выходной день в неделю и свободное посещение школы в дни лечения и обследования в поликлинике (при пульмонологическом центре), освобождение от экзаменов. Вопрос о возможности прививок детям, больным М., решается индивидуально.
Больные М. дети с диспансерного учета не снимаются, а по достижении 15-летнего возраста передаются под наблюдение терапевта в поликлинику для взрослых.
Прогноз при М. серьезный и зависит от тяжести течения заболевания, возраста, в котором появились первые симптомы (если заболевание проявилось на первом году жизни, то прогноз хуже), ранней диагностики и целенаправленною лечения. Прогноз во многом определяется тяжестью течения легочного синдрома (при хронической колонизации легких синегнойной палочкой прогноз хуже).
За последние годы в связи с улучшением диагностики и более ранним назначением адекватной терапии увеличилась продолжительность жизни больных. Однако в связи с хроническим течением заболевания больные нуждаются в постоянном наблюдении и лечении. Если в 50-е годы 80% больных умирали в возрасте до 10 лет, то в 80-е годы летальность составляет около 36% (большинство умерших в возрасте до года). Чем раньше поставлен диагноз, начато целенаправленное лечение и проводится профилактика рецидивов болезни, тем благоприятнее прогноз.
Библиогр.: Врожденные и наследственные заболевания легких у детей, под ред. Ю.Е. Вельтищева и др., с. 170, М., 1986; Рейдерман М.И. Муковисцидоз, М., 1974, библиогр.; Фадеева М.А. Дифференциальная диагностика и принципы лечения муковисцидоза у детей. Вопр. охр. мат. и дет., т. 29, № 11, с. 3, 1984.
Рис. 5. Бронхограмма ребенка 6 лет, больного смешанной формой муковисцидоза: деформированные бронхи, рассеянные цилиндрические и мешотчатые бронхоэктазы.
 Окраска гематоксилином и эозином; ×80″>
Окраска гематоксилином и эозином; ×80″>
Рис. 1. Микропрепарат поджелудочной железы при смешанной форме муковисцидоза: 1 — расширенные и заполненные ацидофильным слоистым секретом междольковые и внутридольковые протоки; 2 — разрастания соединительной ткани. Окраска гематоксилином и эозином; ×80.
 рентгенограмма грудной клетки (прямая проекция) ребенка 3 месяцев, больного смешанной формой муковисцидоза: эмфизема, пневмосклероз, инфильтративный процесс в верхней доле правого легкого»>
рентгенограмма грудной клетки (прямая проекция) ребенка 3 месяцев, больного смешанной формой муковисцидоза: эмфизема, пневмосклероз, инфильтративный процесс в верхней доле правого легкого»>
Рис. 2. Обзорная рентгенограмма грудной клетки (прямая проекция) ребенка 3 месяцев, больного смешанной формой муковисцидоза: эмфизема, пневмосклероз, инфильтративный процесс в верхней доле правого легкого.
Рис. 3. Ребенок с муковисцидозом.
 фаза ремиссии): эмфизема, диффузный пневмосклероз, особенно выраженный в прикорневых зонах»>
фаза ремиссии): эмфизема, диффузный пневмосклероз, особенно выраженный в прикорневых зонах»>
Рис. 4. Обзорная рентгенограмма грудной клетки (прямая проекция) ребенка 7 лет, больного легочной формой муковисцидоза (фаза ремиссии): эмфизема, диффузный пневмосклероз, особенно выраженный в прикорневых зонах.
- ИНВИТРО
- Библиотека
- Справочник заболеваний
- Муковисцидоз…
Муковисцидоз (кистозный фиброз)
Муковисцидоз: причины появления, симптомы, диагностика и способы лечения.
Определение
Муковисцидоз – это мультисистемное, наследственное, опасное для жизни заболевание, характеризующееся поражением экзокринных желез, а также жизненно важных органов и систем: дыхательных путей, желудочно-кишечного тракта, поджелудочной железы, печени, слюнных и потовых желез, репродуктивной системы.
В России частота заболевания составляет 1 на 9000 новорожденных. В прошлом (из-за высокой ранней смертности) муковисцидозом болели в основном дети. Современный уровень диагностики и лечебных мероприятий позволяет значительно продлить жизнь таких больных – до 35-50 лет. В Москве медиана выживаемости достигает 39,5 лет.
Причины появления муковисцидоза
Причиной патологических изменений при муковисцидозе является мутация гена, находящегося в длинном плече 7-й хромосомы и передающегося по аутосомно-рецессивному типу при наследовании двух мутантных генов родителей. Ген назван муковисцидозным трансмембранным регулятором (МВТР). На сегодняшний день выделено более 2000 мутаций этого гена. Он регулирует транспорт электролитов (главным образом хлора) через мембраны эпителиальных клеток, выстилающих выводные протоки экзокринных желез. Мутация приводит к нарушению структуры и функции синтезируемого белка, в результате чего секрет, выделяемый этими железами, становится чрезмерно густым и вязким.
Название заболевания происходит от латинских слов mucus – «слизь» и viscidus – «вязкий».
В патогенезе данного генетического заболевания выделяют следующие патологические звенья:
- нарушение функции экзокринных желез;
- нарушение электролитного обмена;
- поражение соединительной ткани, что часто связано с формированием иммунных комплексов к синегнойной палочке.
Классификация заболевания
В современной классификации выделяют:
- Классический муковисцидоз:
- с панкреатической недостаточностью (смешанная или легочно-кишечная форма заболевания);
- с ненарушенной функцией поджелудочной железы (преимущественно легочная форма заболевания);
- неуточненный – неопределенный диагноз при положительном неонатальном скрининге на муковисцидоз.
- Атипичный муковисцидоз протекает в одной из форм: хронический панкреатит, изолированная обструктивная азооспермия, диссеминированные бронхоэктазы, аллергический бронхолегочный аспергиллез, диффузный панбронхиолит, склерозирующий холангит.
Симптомы муковисцидоза
Тяжесть протекания муковисцидоза напрямую зависит от возраста, когда стали проявляться первые симптомы, — чем младше ребенок, тем тяжелее течение заболевания и неблагоприятнее прогноз. У большинства детей отмечаются часто рецидивирующие респираторные инфекции с явлениями бронхита. Достаточно рано появляется кашель с выделением гнойной мокроты, тошнотой, рвотой и нарушением сна. Также может отмечаться свистящее дыхание, одышка, задержка прибавки веса, частый, с примесью жира и зловонным запахом стул.
Кожные покровы имеют «соленый» привкус в связи с отложением кристаллов соли на коже ребенка.
Симптомы в разные возрастные периоды имеют свои особенности:
- у новорожденных к основным симптомам относят рвоту с примесью желчи, отсутствие стула, вздутие живота, продолжительную обструктивную желтуху, кишечную (мекониевую) непроходимость, выраженный сосудистый рисунок на коже живота, интоксикацию, полигиповитаминоз, быстрое сморщивание кожи пальцев в воде, бронхообструктивный синдром, гипонатриемию и гипокалиемию, анемию, выбухание родничка и паралич лицевого нерва, отставание в физическом развитии, недостаточность ферментативной активности желудочно-кишечного тракта (особенно ярко проявляется после перевода ребенка на искусственное вскармливание или прикорм), быстро развивающуюся гипотрофию;
- у детей дошкольного возраста преобладают бронхообструктивный синдром, рецидивирующие синуситы, полипы носа, непереносимость жары, выпадение прямой кишки, панкреатит, гепатомегалия и др.;
- у детей школьного возраста в мокроте выделяют синегнойную палочку, бронхоэктазы, симптом «барабанных палочек», нарушение углеводного обмена, наблюдаются полиурия, полидипсия, снижение массы тела, цирроз печени, гипертензия, спленомегалия, асцит, варикозное расширение вен пищевода, задержка физического развития.
 У заболевших в раннем детстве и доживших до взрослого возраста отмечаются следующие симптомы: постоянно рецидивирующие инфекционно-воспалительные процессы в легких, изменения бронхиальной стенки, бронхоэктазы, пневмофиброз, буллезная и обструктивная эмфизема, цирротические изменения, сахарный диабет, синуситы и др.
У заболевших в раннем детстве и доживших до взрослого возраста отмечаются следующие симптомы: постоянно рецидивирующие инфекционно-воспалительные процессы в легких, изменения бронхиальной стенки, бронхоэктазы, пневмофиброз, буллезная и обструктивная эмфизема, цирротические изменения, сахарный диабет, синуситы и др.
 Альвеолярный мешок здорового человека (слева) и альвеолярный мешок пациента с муковисцидозом (справа)
Альвеолярный мешок здорового человека (слева) и альвеолярный мешок пациента с муковисцидозом (справа)
Взрослые с поздней манифестацией и атипичной формой заболевания имеют скрытые формы болезни, которые чаще всего случайно проявляются в виде рецидивирующего бронхита, синусита, хронической обструктивной болезни легких, цирроза печени, бесплодия.
Диагностика муковисцидоза
Диагностика муковисцидоза основана на данных лабораторных и инструментальных исследований, клинической картине, физикальном обследовании и тестах функциональной диагностики.
Скрининг новорожденных на муковисцидоз направлен на доклиническое выявление патологии и основан на определении иммунореактивного трипсина в сухом пятне капиллярной крови.
Образец крови берется у доношенного ребенка из пятки на 4-й день жизни через 3 часа после кормления и на 7-й день – у недоношенного.
К специфическим лабораторным критериям муковисцидоза относятся:
- потовый тест — определение электролитов пота (показывает концентрацию ионов натрия и/или хлора);
- исследование назальных потенциалов;
- исследование панкреатической эластазы кала;
Муковисцидоз, CFTR ч.м.
Исследование частых мутаций в гене CFTR.
Тип наследования.
Аутосомно-рецессивный.
Гены, ответственные за развитие заболевания.
CFTR -трансме…
Основные наследственные заболевания
Определение носительства частых мутаций в генах, ответственных за развитие наиболее частых аутосомно-рецессивных заболеваний: муковисцидоз, несиндромальная ней�…
Основными инструментальными методами диагностики муковисцидоза являются:
- рентгенография;
- исследование функции внешнего дыхания;
Эхокардиография
Исследование, позволяющее оценить функциональные и органические изменения сердца, его сократимость, а также состояние клапанного аппарата.
Если ребенку поставлен диагноз «муковисцидоз», то генетический анализ потребуется всем членам семьи. Это важно не только для подтверждения диагноза, но и для дородовой диагностики при последующих беременностях.
Дифференциальная диагностика муковисцидоза проводится с туберкулезом, мекониальным илеусом, желтухой новорожденных, гемолитической анемией, выпадением прямой кишки, рецидивирующими бронхитом, синуситом, бронхиальной астмой, бронхоэктазами и другими заболеваниями.
К каким врачам обращаться
Если у ребенка наблюдаются какие-то из перечисленных симптомов, следует безотлагательно сообщить об этом
врачу-педиатру
. При подозрении на муковисцидоз участковый педиатр дает направление в Российский или региональный центр по борьбе с муковисцидозом. Лечение муковисцидоза представляет собой трудную задачу, которая требует консультаций
пульмонолога
,
гастроэнтеролога
и других специалистов.
Лечение муковисцидоза
Терапия муковисцидоза носит комплексный характер и направлена на разжижение и удаление вязкой мокроты из бронхов, борьбу с инфекционными заболеваниями легких, замещение недостающих ферментов поджелудочной железы, коррекцию поливитаминной недостаточности, разжижение желчи.
Важное место в терапии бронхо-легочной формы муковисцидоза занимает кинезотерапия (специальный комплекс упражнений и дыхательной гимнастики, направленных на удаление мокроты). Занятия должны быть ежедневными и пожизненными. Необходимо обеспечить физическое развитие ребенка согласно возрастным нормам, максимально высокое качество жизни пациента, профилактику осложнений.
В зависимости от тяжести состояния лечение может проводиться в специализированном отделении больницы, в дневных стационарах или на дому. Такое лечение включает:
- проведение активной муколитической терапии;
- назначение бронхолитической терапии (ингаляционных бронходилататоров, в некоторых случаях кортикостероидов);
- антибактериальную терапию, включая ингаляционную;
- противогрибковую терапию (по показаниям);
- лечение гепатотропными препаратами;
- высококалорийную диету (иногда требуется дополнительное энтеральное зондовое питание).
Хирургические методы лечения муковисцидоза:
- лапаротомия с резекцией части кишки (в период новорожденности при мекониальном илеусе);
- полипэктомия (при полипозе носа);
- постановка порт-системы (венозного катетера, имплантируемого под кожу) — при длительных курсах внутривенной антибактериальной терапии;
- спленэктомия, шунтирующие и склерозирующие манипуляции (при портальной гипертензии);
- плеврэктомия, плевродез (при повторных пневмотораксах).
При тяжелом течении заболевания необходимо пребывание пациента в условиях стационара с длительной кислородотерапией и практически постоянный прием антибактериальных препаратов.
При соблюдении этих условий терминальная фаза болезни может продолжаться несколько лет. В этой фазе больные планируются на пересадку легких.
Очень важную роль в лечении детей с муковисцидозом играют родители. На их плечи ложится большая ответственность по уходу за больным ребенком, поскольку терапия этого заболевания пожизненная и требует скрупулезного выполнения всех рекомендаций врача. Только родители, постоянно находясь с ребенком, могут оценить изменение в состоянии малыша и вовремя обратиться за медицинской помощью.
Осложнения
Основной причиной осложнений и летальности являются патологии дыхательных путей. К неблагоприятным и тяжелым последствиям муковисцидоза относятся:
- абсцессы;
- ателектазы;
- пневмоторакс, пиопневмоторакс;
- гипоксемия;
- гиперкапния;
- легочное сердце;
- желудочное и легочное кровотечение;
- отечный синдром;
- сахарный диабет;
- печеночная энцефалопатия;
- амилоидоз почек;
- острая и хроническая почечная недостаточность;
- фиброз печени, гепатоцеллюлярная недостаточность, желчнокаменная болезнь;
- неукротимая рвота;
- диарея, приводящая к обезвоживанию и выраженным электролитным нарушениям и др.
Рецидивирующие респираторные заболевания в виде бронхитов, бронхиолитов, пневмонии приводят к еще большему увеличению вязкости мокроты, обструкции дыхательных путей, инфекциям и частым воспалениям.
Повышенная вязкость секрета желез внешней секреции опасна хроническим воспалительным процессом в легких, экзокринной недостаточностью поджелудочной железы, гепатобилиарной патологией и аномально высоким содержанием электролитов в поте.
Вязкий кишечный секрет может привести к мекониальной непроходимости у новорожденных (мекониальному илеусу) и иногда к синдрому мекониевой пробки. У детей старшего возраста и взрослых также могут быть симптомы хронического запора и непроходимости кишечника.
Другие проблемы со стороны желудочно-кишечного тракта включают инвагинацию кишечника, странгуляционную кишечную непроходимость, ректальный пролапс, периаппендикулярный абсцесс, панкреатит, повышенный риск развития рака гепатобилиарного и желудочно-кишечного трактов (включая рак поджелудочной железы), гастроэзофагеальный рефлюкс, эзофагит, болезнь Крона и целиакию.
Бесплодие наблюдается у 98% мужчин из-за недоразвития семявыносящих протоков или других форм обструктивной азооспермии. У женщин фертильность несколько снижается из-за вязкого цервикального секрета, хотя многие женщины вынашивают беременность полностью. Исходы беременности для матери и новорожденного связаны со здоровьем матери.
Другие осложнения включают остеопению/остеопороз, депрессию и беспокойство, хроническую боль, обструктивное апное во сне, почечные камни, диализозависимую хроническую почечную недостаточность, железодефицитную анемию и эпизодические артралгии/артриты.
Течение заболевания во многом определяется степенью поражения легких. Ухудшение неизбежно, что приводит к истощению и, в конечном итоге, к смерти, как правило, из-за сочетания дыхательной недостаточности и легочного сердца. Прогрессирование легочной и сердечной недостаточности является наиболее частой причиной смерти пациентов (95%). Среди других причин выделяют осложнения при трансплантации органов (12%), заболевания печени и печеночную недостаточность (2,3%), травмы (2,1%), суицид (0,8%), другие (1,3%).
Профилактика муковисцидоза
Если в семье есть случаи муковисцидоза, то при планировании беременности обязательно следует обратиться к медицинскому генетику. В настоящее время стала возможной дородовая диагностика муковисцидоза у плода. Именно поэтому при возникновении каждой новой беременности необходимо сразу же (не позднее 8 недели беременности) обратиться в центр дородовой диагностики.
В конце ХХ века начали создаваться Российские и региональные центры по борьбе с муковисцидозом. Основу терапевтической помощи больным составляет грамотно подобранная пожизненная лекарственная терапия, регулярные профилактические осмотры и стационарное лечение в период обострений.
Источники:
- Клинические рекомендации «Кистозный фиброз (муковисцидоз)». Разраб.: Союз педиатров России, Ассоциация медицинских генетиков, Российское респираторное общество. – 2019. – 89 с.
- Баранов А.А., Намазова-Баранова Л.С., Симонова О.И., Каширская Н.Ю. и соавт. Современные представления о диагностике и лечении детей с муковисцидозом // Педиатрическая фармакология. – 2015; 12 (5): 589–604.
- Aurora P, Gustafsson P, Bush A, et al: Multiple breath inert gas washout as a measure of ventilation distribution in children with cystic fibrosis. Thorax 59:1068–1073, 2004.
ВАЖНО!
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Информация проверена экспертом

Лишова Екатерина Александровна
Высшее медицинское образование, опыт работы — 19 лет
Поделитесь этой статьей сейчас
Рекомендации
-
2410
05 Марта
-
13068
01 Марта
-
13065
23 Февраля
Похожие статьи
Описторхоз
Описторхоз: причины появления, симптомы, диагностика и способы лечения.
Микоз
Микоз: причины появления, симптомы, диагностика и способы лечения.
Гломерулонефрит
Гломерулонефрит: причины появления, симптомы, диагностика и способы лечения.
Муковисцидоз
![]()
Версия: Клинические протоколы МЗ РК — 2018 (Казахстан)
Категории МКБ:
Кистозный фиброз неуточненный (E84.9), Кистозный фиброз с другими проявлениями (E84.8), Кистозный фиброз с кишечными проявлениями (E84.1), Кистозный фиброз с легочными проявлениями (E84.0)
Разделы медицины:
Педиатрия, Пульмонология, Пульмонология детская
Общая информация
Краткое описание
Одобрен
Объединенной комиссией по качеству медицинских услуг
Министерства здравоохранения Республики Казахстан
от «19» апреля 2019 года
Протокол №63
Муковисцидоз (МВ) (кистозный фиброз) – это аутосомно-рецессивное моногенное заболевание, обусловленное мутацией гена CFTR (трансмембранный регулятор муковисцидоза), характеризующееся поражением экзокринных желез, жизненно важных органов и систем, имеющее тяжелое течение и прогноз.
Мультисистемное генетическое заболевание, связанное с нарушениями транспорта натрия и воды через эпителиальные поверхности. Следствием мутации гена является нарушение синтеза, структуры и функции белка трансмембранного регулятора проводимости муковисцидоза (CFTR), в результате чего хлорные каналы становятся непроницаемыми для ионов хлора при гиперабсорбции натрия и воды, что вызывает дегидратацию апикальной поверхности секреторного эпителия, увеличение вязкости слизи. Поражаются дыхательные пути, желудочно-кишечный тракт, печень, поджелудочная железа, слюнные, потовые железы, репродуктивная система. Патология дыхательной системы — главная причина осложнений и летальности (более чем в 90%). Возраст манифестации заболевания очень различается.
ВВОДНАЯ ЧАСТЬ
Название протокола: Муковисцидоз
Код (ы) МКБ 10:
| МКБ 10 | |
| Код | Название |
| Е 84 | Кистозный фиброз |
| Е84.0 | Кистозный фиброз с легочными проявлениями |
| Е84.1 | Кистозный фиброз с кишечными проявлениями |
| E84.8 | Кистозный фиброз с другими проявлениями |
| E84.9 | Кистозный фиброз неуточненный |
Дата разработки/пересмотра протокола: 2014 год (пересмотр 2019 г.)
Сокращения, используемые в протоколе:
| АБЛА | аллергический бронхолегочный аспергиллез |
| АБП | антибактериальный препарат |
| АБТ | антибактериальная терапия |
| АДР | аутогенный дренаж |
| ААДР | ассистированный аутогенный дренаж |
| АМГ | аминогликозид |
| БЭН | белково-энергетическая недостаточность |
| ВАТ | вибро-акустическая терапия |
| ВОП | врач общей практики ПМСП |
| ВРВП | варикозное расширение вен пищевода |
| ГКС | глюкокортикостероиды |
| ДКТ | длительная кислородотерапия |
| ДН | дыхательная недостаточность |
| ЖЕЛ | жизненная емкость легких |
| ЖКБ | желчнокаменная болезнь |
| ЖКТ | желудочно-кишечный тракт |
| ИМТ | индекс массы тела |
| КТ | компьютерная томография |
| МВ | муковисцидоз |
| МВТР/CFTR | муковисцидозный трансмембранный регулятор |
| МЗСД | муковисцидоз зависимый сахарный диабет |
| МК | максимальная концентрация |
| МПКЦ | минимальная подавляющая концентрация |
| МПК | минеральная плотность кости |
| МРИ | массо-ростовой индекс |
| ОРЗ | острые респираторные заболевания |
| ОАК | общий анализ крови |
| ОФВ1 | объем форсированного выдоха за 1 секунду |
| ОАМ | общий анализ мочи |
| ПЖ | поджелудочная железа |
| СД | сахарный диабет |
| СЦТ | среднецепочечными триглицеридами |
| УД | уровень доказательства |
| УЗИ | ультразвуковое исследование |
| ФЖЕЛ | форсированная жизненная емкость легких |
| ХАЛ | хронический аспергиллез легких |
| ХДН | хроническая дыхательная недостаточность |
| ЭГДС | эзофагогастродуоденоскопия |
| ЭКГ | электрокардиограмма |
| ЭПН | экзокринная панкреатическая недостаточность |
| ЭхоКГ | эхокардиография |
| РаО2 | парциальное напряжение кислорода |
| РаСО2 | парциальное напряжение углекислого газа |
| РЕР | Positive Expiratory Pressure, положительное давление на выдохе |
| SаO2 | сатурация, насыщение крови кислородом |
Пользователи протокола: педиатры, пульмонологи, врачи общей практики, врачи скорой и неотложной медицинской помощи, терапевты, реабилитологи, диетологи, эндокринологи, хирурги, отоларингологи, трансплантологи.
Категория пациентов: взрослые, дети.
Шкала уровня доказательности (УД):
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортных или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+). Результаты, которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GPP | Наилучшая фармацевтическая практика. |
Облачная МИС «МедЭлемент»
Облачная МИС «МедЭлемент»

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место — 800 RUB / 5500 KZT / 27 BYN в месяц
Классификация
С учетом имеющегося опыта к практическому применению рекомендована следующая классификации МВ (табл.1):
Таблица 1 — Клиническая классификация муковисцидоза:
| Форма болезни | Характеристика бронхолегочных изменении | Другие проявления заболева-ния | Осложнения | ||
| Клиническая | Фаза, активность процесса | Степень ДН ** | |||
|
1.Смешанная или легочно — кишечная форма заболевания (муковисцидоз с панкреатичес-кой недостаточностью)
|
1.Хронический обструктивный бронхит.
|
1. Ремиссия.
|
0 I ст. II ст. III ст |
— Синусит — Синдром псевдо — Барттера — Азоо-спермия -Рецидиви-рующий панкреатит |
Абсцессы, ателектазы, пневмо-пиопневмоторакс, кровохарканье, кровотечение (легочное, желудочное), аллергический бронхолегочный аспергиллез (АБЛА). Полипоз носа Мекониевый илеус, эквиваленты мекониевого илеуса, выпадение прямой кишки, Цирроз печени (без и с портальной гипертензией) ЖКБ Отставание в физическом развитии. Белково-энергетическая недостаточность Нарушение толерантности к углеводам Муковисцидоз-ассоции-рованный сахарный диабет Снижение минеральной плотности костной ткани Вторичный остеопороз Амилоидоз почек Сиалоаденит Витамин К-дефицитные состояния (геморрагическая болезнь Дефицитные состояния (анемия и др.) Легочная гипертензия, Сердечно-сосудистая недостаточность |
| Генотип (мутации гена CFTR) | |||||
|
Микробиологический статус (указывается дата первичного высева микробного патогена (патогенов) и, если есть, последнего) |
Стафилококковая инфекция. Синегнойная инфекция Инфекция, вызванная B. cepacia Другие инфекции Микробные ассоциации |
||||
|
Другие формы: Неопределенный диагноз при положительном неонатальном скрининге на муковисцидоз*** Заболевания, связанные с геном CFTR****: – изолированная обструктивная азооспермия; – хронический панкреатит; – диссеминированные бронхоэктазы |
Примечания:
** Степень дыхательной недостаточности устанавливается согласно «Классификации дыхательной недостаточности». Степень тяжести заболевания рекомендуется не указывать исходя из первично-хронического течения, полиорганного поражения и прогредиентного течения.
*** Положительный неонатальный скрининг или неонатальная гипертрипсиногенемия (см. раздел «Диагностика муковисцидоза. Неонатальный скрининг») не являются диагнозом и в классификацию не включены, пациентам с неонатальной гипертрипсиногенемией рекомендуется в 1 год провести повторно потовую пробу. Предложен новый диагноз – «неопределенный диагноз при положительном неонатальном скрининге на муковисцидоз» (см. Раздел «Генетика муковисцидоза»).
**** Код МКБ рекомендуется использовать из соответствующих разделов.
Основное осложнение МВ, причина инвалидизации и смертности — дыхательная недостаточность (ДН).
Таблица 2 — Классификация дыхательной недостаточности по степени тяжести.
| Степень | РаО2, мм рт. ст. | SаO2, % | РаСО2 |
| Норма | > 80 | > 95 | 36-44 |
| I | 70—79 | 90—94 | <50 |
| II | 50—69 | 75—89 | 50-70 |
| III | <50 | <75 | > 70 |
Примеры формулировки диагноза:
— Муковисцидоз [генотип: гетерозигота по мутации 2143delT], легочно-кишечная форма, тяжелое течение. Распространенные цилиндрические бронхоэктазы обоих легких, обострение. ДН II ст. Хронический полипозно-гнойный пансинусит. Назальный полипоз 2 ст. Хроническая панкреатическая недостаточность, тяжелая степень. Дуоденит. Бульбит. Дуоденогастральный рефлюкс. Цирроз печени (F4 по шкале METAVIR — по данным фиброэластометрии печени). Нарушение толерантности к глюкозе.
— Муковисцидоз (генотип гетерозигота по мутации гена F508 Del 21kb), смешанная форма. Двусторонняя кистозно-буллезная деформация легких, множественные бронхоэктазы, распространенная локализация, осложненное течение. Хроническое носительство Pseudomonas aeruginosa. ДН 2 ст. Хроническая панкреатическая недостаточность. Снижение нутритивного статуса.
Диагностика
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ [1-12,17,33, 36-41, 72]
Диагностические критерии [1-12]:
В большинстве случаев МВ диагностируется в раннем детском возрасте (90% случаев – на первом году жизни). Нередки случаи диагностики МВ у взрослых с классическим фенотипом (хронический/рецидивирующий бронхит, синусит, панкреатит).
Классический фенотип является следствием двух мутантных копий гена муковисцидозного трансмембранного регулятора (CFTR), и проявляется хронической бактериальной инфекцией дыхательных путей, придаточных пазух носа, стеатореей вследствие внешнесекреторной недостаточности поджелудочной железы, мужским бесплодием из-за обструктивной азооспермии, повышенной концентрацией хлоридов потовой жидкости.
Диагностические сложности при МВ обусловлены фенотипическим разнообразием в связи с генетическим полиморфизмом. Пациенты с атипичным МВ имеют как минимум одну копию мутантного гена CFTR, функция которого частично сохранена («мягкие» мутации). Это приводит к диагностике МВ во взрослом возрасте, у таких пациентов отмечается более мягкое течение болезни в связи с сохранностью функции поджелудочной железы и нетяжелым поражением органов дыхания.
Жалобы:
- мучительный приступообразный кашель;
- трудноотделяемая гнойная вязкая мокрота;
- свистящее дыхание;
- нарушение носового дыхания (заложенность);
- гнойные выделения из носа;
- одышка (затрудненное дыхание);
- боли в животе, обильный, частый (4-6 раз в сутки), блестящий, жирный, зловонный стул;
- слабость, утомляемость, задержка развития;
- снижение массы тела/задержка в прибавке веса.
Анамнез:
- данные семейного анамнеза о смерти детей на первом году жизни или наличие сибсов со сходными клиническими проявлениями;
- мекониальный илеус и его эквиваленты;
- синдром нарушенного кишечного всасывания неясного генеза;
- учащенный стул со зловонным запахом;
- признаки хронического синусита с поражением всех синусов, полипы носа, особенно в раннем возрасте;
- рецидивирующие бронхиты, бронхиолиты;
- повторные и рецидивирующие пневмонии с затяжным течением;
- частые курсы антибактериальной терапии для лечения кашля;
- высев Ps.aeruginosa;
- желтуха обструктивного типа у новорожденных с затяжным течением;
- бронхиальная астма, рефрактерная к традиционной терапии;
- рецидивный панкреатит и/или аппендицит
- бронхоэктазии;
- у более старших пациентов (мужчин) бесплодие вследствие врожденного билатерального отсутствия семявыводящего протока;
- циррозы печени;
- сахарный диабет с респираторным синдромом;
- гастроэзофагеальный рефлюкс;
- холелитиаз;
- выпадение прямой кишки;
- задержка полового развития.
Физикальное обследование:
- нарушение роста и развития: снижение массы тела;
- могут наблюдаться различные деформации грудной клетки
- при типичном течении отмечается характерный внешний вид: «кукольное» лицо, расширенная, деформированная грудная клетка бочкообразной формы с выбуханием грудины, большой, вздутый, иногда «лягушачий живот», худые конечности;
- соленый вкус кожи;
- при распространенном поражении легочной ткани признаки хронической гипоксии: деформации концевых фаланг пальцев по типу «барабанных пальцев» и/или ногтей по типу «часовых стекол»;
- перкуторно над легкими может выслушиваться коробочный звук и/или участки притупления,
- при аускультации – ослабление дыхания, сухие и разнокалиберные (преимущественно среднепузырчатые) влажные хрипы локальные или распространенные, в зависимости от объема поражения.
- при обследовании верхних дыхательных путей выявляется хронический пансинусит, нередко полипозный;
- периферические отеки;
- сахарный диабет в сочетании с респираторными симптомами;
- гепатомегалия, спленомегалия (при развитии цирроза печени).
Манифестация клинических проявлений МВ (начальные признаки) может протекать по-разному:
- мекониальный илеус — 15%;
- респираторные симптомы — 25%;
- кишечные проявления — 30%;
- респираторные и кишечные — 15%;
- отягощенный семейный анамнез по МВ — 10%;
- другие проявления — 5%.
В различные возрастные периоды имеются клинические особенности МВ (табл.3).
Таблица 3 — Клинические проявления МВ в разных возрастных категориях:
| Возраст | Симптомы и синдромы |
| Грудной возраст |
Рецидивирующие или хронические респираторные симптомы (кашель, одышка) Рецидивирующие бронхиты/бронхиолиты Плохая прибавка веса Повторные пневмонии/эмпиема Затяжная желтуха новорожденных Неоформленный, обильный, маслянистый и зловонный стул, стеаторея Мекониевый илеус, хроническая диарея Выпадение прямой кишки Отставание в физическом развитии Гипопротеинемические отеки Соленый вкус кожи Синдром псевдо-Барттера Повышенная кровоточивость, связанная с дефицитом витамина K Данные семейного анамнеза о смерти детей на первом году жизни или наличие сибсов со сходными клиническими проявлениями |
| Дошкольный |
Стойкий кашель с/без гнойной мокроты; рецидивирующая/хроническая одышка Рецидивирующая инфекция органов дыхания или астма Идиопатические бронхоэктазы Отставание в весе и росте Выпадение прямой кишки, инвагинация Хроническая диарея, стеаторея Симптом «барабанных палочек» Кристаллы соли на коже Гипотоническая дегидратация Гипоэлектролитемия и метаболический алкалоз Гепатомегалия или диагностически неясное нарушение функции печени Синуситы и назальный полипоз Тепловой удар с гипонатриемией |
| Школьный |
Рецидивирующая инфекция органов дыхания или астма Pseudomonas aeruginosa в мокроте Хронический синусит Назальный полипоз Идиопатические бронхоэктазы Симптом «барабанных палочек» Стеаторея, Хроническая диарея Синдром дистальной интестинальной обструкции Панкреатит Выпадение прямой кишки Нарушение толерантности к углеводам / Сахарный диабет Гепатомегалия Заболевание печени неясной этиологии Фокальный билиарный цирроз Хроническая интестинальная обструкция, инвагинация Тепловой удар с гипонатриемией |
| Подростки и взрослые |
Гнойное заболевание легких неясной этиологии Бронхоэктазы Хронический синусит Симптом «барабанных палочек» Аллергический бронхолегочный аспергиллез Острый или хронический панкреатит Синдром дистальной интестинальной обструкции Нарушение толерантности к углеводам/Сахарный диабет в сочетании с респираторными симптомами Отставание в росте Задержка полового развития Азооспермия/двусторонняя атрезия семявыносящих протоков у лиц мужского пола Снижение фертильности у лиц женского пола Фокальный билиарный цирроз Портальная гипертензия Холелитиаз |
Особенности клинической картины муковисцидоза у взрослых.
МВ взрослых делится на две группы:
- больные с «типичной» формой заболевания, заболевшие в раннем детстве и дожившие до взрослого возраста;
- больные с атипичной формой заболевания, с поздней манифестацией.
Первая группа
характеризуется низким нутритивным статусом, непрерывно-рецидивирующим течением инфекционно-воспалительного процесса в легких на фоне выраженных изменений бронхиальной стенки, массивных бронхиоло- и бронхоэктазов, пневмофиброза, эмфиземы. Дыхательные пути часто инфицированы грамотрицательной микрофлорой: Ps.aeruginosa, B.cepacia, Stentrophomonas maltofilia, Alcaligenes xylosoxidans. Отмечается обструктивный тип вентиляционных нарушений, легочная гипертензия. Наблюдается высокая частота «мягких» генотипов с меньшей выраженностью панкреатической недостаточности; цирротические изменения, пансинуситы, кровохарканье, сахарный диабет (20%) и другие легочные и внелегочные осложнения.
Cтертые формы (при мягкой мутации в гене CFTR): диагностируется часто во взрослом возрасте, под разными «масками»: синусит, рецидивирующий бронхит, хронические обструктивные болезни легких, цирроз печени, мужское бесплодие.
Тяжесть заболевания зависит от сроков появления первых симптомов: чем младше ребенок на момент манифестации МВ, тем тяжелее течение и неблагоприятнее прогноз. Почти у всех больных МВ мужского пола (97%) — азооспермия, связанная с врожденным отсутствием, атрофией или обструкцией семенного канатика. Большинство мужчин с МВ не способны иметь потомство. У пациентов женского пола заболевание сопровождается снижением фертильности (20%), но у большинства детородная функция сохранена.
Лабораторные исследования [1, 2, 9, 17, 33, 36-41, 72]:
- определение хлоридов в потовой жидкости (проба Минора) — наиболее информативный диагностический тест. Потовую пробу можно проводить детям любого возраста. При недостаточном образовании пота необходимо повторное исследование через неделю. Тест положительный при наличии хлоридов пота >60 ммоль/л [≥ 60 мэкв/л]. Отрицательная потовая проба (при значении хлоридов пота <30 ммоль/л [<30 мэкв/л] во всех возрастных группах) указывает, что муковисцидоз маловероятен. Положительная потовая проба свидетельствует о наличии муковисцидоза. При результатах потовой пробы со уровнем хлоридов пота 30-59 ммоль/л [30-59 мэкв/л], требуются дополнительные исследования – молекулярно-генетический анализ (УД – А) [1, 2, 9, 17, 33, 72].
- Молекулярно-генетический анализ гена МВТР (CFTR) [1, 2, 9, 17, 33, 72]: диагностическим подтверждением МВ является выявление двух патогенных мутации в гене CFTR в трансположении, вызывающие муковисцидоз (УД А). Молекулярно-генетический анализ обязательно показан при:
— пограничных результатах потовой пробы;
— невозможности проведения потовой пробы (недостаточный вес, незрелость новорожденного, тяжесть состояния, др.);
— при неонатальной гипертрипсиногенемии и отрицательном результате потовой пробы;
NB! учитывая современные подходы к патогенетической терапии МВ, всем пациентам с МВ должно быть рекомендовано проведение генетического исследования.
- Микробиологическое исследование мокроты. Микробная флора обычно представлена ассоциациями (S. аureus, Pseudomonas aeruginosa, Burkholderia cepacia complex, Achromobacter spp, Stenotrophomonas maltophilia, Acinetobacter, микромицеты и др.). Для исследования кроме мокроты можно учитывать мазок из зева, назофарингеальный аспират, бронхоальвеолярный лаваж (УД А) [36-41].
- определение уровня панкреатической эластазы в кале для определения панкреатической недостаточности; хроническая панкреатическая недостаточность средней степени тяжести — уровень эластазы от 100 до 199 мг/г; тяжелой степени — <100 мг/г; крайне тяжелая – ниже 15 мг/г (УД А) [1, 2, 9, 17, 33, 72]. Исследование проводится через 3 месяца на первом году жизни, далее ежегодно в детском возрасте, в периоды замедления роста, потери веса и диареи.
- исследование копрограммы с определением уровня нейтрального жира (УД А) [1, 2, 9, 17, 33, 72]: степени выраженности стеатореи I типа: выраженная – визуально жирный стул; умеренная – визуально жира нет, в копрограмме нейтральный жир в повышенном количестве; скрытая – визуально жира нет, нейтральный жир в копрограмме повышен незначительно.
- общий анализ крови (может быть снижение Нb, лейкоцитоз/лейкопения, тромбоцитоз/тромбоцитопения, увеличение СОЭ)
- биохимический анализ крови с определением: билирубина (общего и прямого), аланинаминотрансферазы (АЛаТ) и аспартатаминотрансферазы (АСаТ) (часто повышение этих показателей), глюкозы (часто повышение), общего белка и альбумина (часто снижение);
- определение С-реактивного белка (увеличен вследствие наличия воспалительного процесса);
- Иммунограмма + иммуноглобулины А, М, G, Е (часто снижены);
- определение уровня железа (Fe), ферритина, В12, фолатов, витамина Д (25(OH)D3), церулоплазмина в сыворотке крови (у большинства пациентов снижены);
- определение содержания электролитов (натрий, калий, кальций ионизированные) в сыворотке крови (как правило снижены);
- коагулограмма с определением аутокоагуляционного теста, активированного времени рекальцификации (АВР), активированного частичного тромбопластинового времени (АЧТВ), тромбинового времени (ТВ) (признаки ДВС-синдрома и гнойно-воспалительных изменений);
- общий анализ мочи (могут быть протеинурия, лейкоцитурия, ураты);
- диагностика остеопении/остеопороза: кальций общий, ионизированный, электролиты, 25(OH)D3, щелочная фосфатаза, фосфор органический, остеокальцин, кальцитонин, паратгормон, beta-cross laps в крови; маркеры костного обмена: P1NP (маркер костеообразования) и СТХ (маркер резорбции кости);
- лабораторные исследования на аллергический бронхолегочный аспергиллез: уровень общего иммуноглобулина Е (IgE), специфические IgE и IgG к Aspergillus fumigatus, возможно проведение кожного тестирования с антигеном Aspergillus fumigates.
Инструментальные исследования:
- КТ органов грудной клетки: кистозные изменения, бронхоэктазии (УД – В).
- Рентгенография органов грудной клетки (может выявить деформацию и усиление легочного рисунка, пневмофиброз, перибронхиальную инфильтрацию, ателектаз), данный метод недостаточно информативен при муковисцидозе.
- Рентгенография пазух носа (может помочь выявить пансинусит);
- Пульсоксиметрия – определение насыщения крови кислородом (отмечается снижение периферической сатурации;
- УЗИ органов брюшной полости: диффузные изменения поджелудочной железы, печени, кистофиброз, изменения размеров; особое внимание обращают на наличие кист в поджелудочной железе тип кровотока в печени. Наличие линейного кровотока в печени свидетельствует о формировании фиброза.
- Спирометрия (взрослым и детям старше 5 лет). Наиболее часто у пациентов с МВ выявляются обструктивные и рестриктивные нарушения вентиляции (в зависимости от объема и характера поражения бронхиального дерева).
- Пикфлоуметрия (детям старше 5 лет): для контроля состояния бронхиальной проходимости (в случае невозможности проведения спирометрии).
- ЭКГ (в 12 отведения);
- ЭхоКГ с допплеровским анализом (измерение градиента давления на легочной артерии) проводится регулярно для диагностики легочной гипертензии и легочного сердца.
- ФЭГДС: для определения состояния слизистой отделов желудочно-кишечного тракта, исключения/подтверждения признаков портальной гипертензии.
- Исследование газового состава артериальной крови при снижении периферической сатурации до 90%.
Показания для консультации специалистов:
- консультация диетолога – для коррекции питания;
- консультация гастроэнтеролога, гепатолога – для коррекции желудочно-кишечных нарушений и нарушении функции печени;
- консультация пульмонолога – для диагностики и коррекции бронхолегочных изменений;
- консультация генетика – для медико-генетического консультирования и проведения молекулярно-генетического анализа гена CFTR;
- консультация реабилитолога – для подбора и объема легочной и общей реабилитации;
- консультация психолога – для оказания психологической помощи как самому пациенту, так и членам семьи/опекунам;
- консультация оториноларинголога – для санации очагов хронической инфекции
- консультация эндокринолога – при нарушении углеводного обмена, остеопении/остеопорозе;
- консультация стоматолога – для санации очагов хронической инфекции ротовой полости;
- консультация трансплантолога – при необходимости трансплантации легких, печени.
- консультации остальных специалистов по мере необходимости.
Диагностический алгоритм [31]: (схема-1)


Дифференциальный диагноз
Дифференциальный диагноз и обоснование дополнительных исследований [9,17]:
Перечень заболеваний и состояний, требующих дифференциальной диагностики с МВ, отличается в зависимости от возрастной категории пациента. Наиболее частые нозологические формы для дифференциального диагноза при МВ:
- врожденные аномалии бронхиального дерева (КТ грудного сегмента);
- первичные или приобретенные иммунодефицитные состояния (определение уровней иммуноглобулинов G, M, A, Е; по показаниям, субклассов иммуноглобулинов, уровня и функции T-, В-клеток, фагоцитоза, системы комплемента, естественных киллеров, тесты на ВИЧ;
- первичная цилиарная дискинезия (характерные клинические проявления: триада Картагенера (хронический бронхит, хронический синусит, обратное расположение внутренних органов), микроскопия биоптата слизистой оболочки носа и/или бронха).
Перечень дополнительных исследований для дифференциальной диагностики и диагностики осложнений:
- КТ придаточных пазух носа: при наличии клинических признаков пансинусита и неинформативности рентгенографии пазух носа.
- Фиброэластометрии печени (фиброскан) при клинико-инструментальных признаках фиброза/цирроза печении с целью оценки степени выраженности фиброза по шкале METAVIR.
- Денситометрия при наличии клинических и/или лабораторных признаках остеопении/остеопороза;
- Фибробронхоскопия: показания ограничены (в связи с наличием других неинвазивных методов диагностики и риска инфекционных осложнений), проводится при необходимости дополнительного микробиологического исследования бронхоальвеолярного лаважа, при легочном кровотечении для установки источника и возможной эмболизации. «Санационные ФБС» не показаны в связи со значительным нарушением мукоцилиарного транспорта после их проведения и отсутствием доказательств их эффективности;
- Рентгенологическое исследование желудочно-кишечного тракта: Ирригоскопияирригография (двойное контрастирование) при осложнении со стороны ЖКТ.
Таблица 4 – Дифференциальная диагностика муковисцидоза:
| Признаки | Муковисцидоз | Астма | Целиакия | Врожденые пороки легких |
| Начало заболевания | Вскоре после рождения | Позже | Чаще после 6 мес., до 2-3 лет | Чаще в период новорожденности и в первые месяцы жизни |
| Масса тела при рождении | Часто низкая | Нормальная | Нормальная | Средняя/ Ниже средней |
| Семейная предрасположенность | Часто подобное заболевание у двоюродных братьев и сестер | Наследственная отягощенность по аллергии, атопии. |
Иногда наблюдается у родителей |
Нет |
| Акушерский анамнез | Отягощенный акушерский анамнез: мертворождение, выкидыши, наличие больного с МВ в семье | Без особенностей | Без особенностей | Интеркуррентные заболевания матери в первом триместре беременности |
| Заболевания органов дыхания | Тяжелые поражения бронхолегочной системы, трудно поддающиеся лечению с момента рождения |
Внезапно, связано с экспозицией аллергена. Быстрое облегчение симптомов самостоятельно или под воздействием терапии |
Может быть вялотекущая пневмония, поддающаяся комплексному лечению | Характерно, поддается лечению |
| Аппетит | Хороший, повышен | Не изменен | Снижен | Не изменен |
| Поражение печени | Часто | Не характерно | Не характерно | Не характерно |
| Гипотрофия | С первых месяцев жизни, постепенно нарастая до II- III степени | Не характерно | Обычно со второго полугодия, быстро прогрессирует | Редко |
| Соленый привкус кожи | Характерно | Не характерно | Не характерно | Не характерно |
| Симптом «барабанных палочек» | Чаще в раннем возрасте | Не характерно | Не характерно | Развивается позже |
| Неврологический статус | Без отклонений | Без отклонений | Раздражительность, мышечная гипотония, судороги | Без отклонений |
| Лабораторно-диагностический тест |
Повышение уровня хлоридов в поте, стеаторея с преобладанием нейтрального жира |
Повышение Ig Е |
Нарушение всасывания углеводов, жиров, белков, повышение IgA в крови | Не характерно |
| Белковый обмен | Гипопротеинемия | В норме |
Тяжелая гипопротеинемия |
В норме |
| IgA, Ig G, Ig M | В норме | В норме | Повышение IgA | В норме |
| Исследование кала | Жидкий, светло-желтый, глинистый, жирный, «зловонный» | Без особенностей | Обильный, разжиженный, светло-желтый, гнилостный | Без особенностей |
| Нейтральный жир | В большом количестве | Отсутствует |
В небольшом количестве |
Не характерен |
| Трипсин | Резко снижен до полного отсутствия | В норме | Умеренно снижен | Нормальный |
| Мутации гена CFTR | Да | Нет | Нет | Нет |
| Хлориды в потовой жидкости | Повышены | В норме | В норме | В норме |
|
Рентгенологическое исследование грудной клетки и
|
Деформация бронхолегочного рисунка, ателектазы, пневмофиброз, бронхоэктазы в ранние сроки болезни | Признаки эмфиземы в поздних стадиях | Без особенностей | Подвижность и пролабирование задней стенки трахеи, признаки гипоплазии |
| Дискинезия тонкой кишки, рельеф слизистой оболочки грубый, «спикулы» псевдодивертикул, большое количество слизи в кишечнике | Без особенностей | Расширение петель кишечника, гипотония, дискинезия кишечника, горизонтальные уровни жидкости | Без особенностей | |
| Спирография | Смешанный тип нарушения вентиляции | Обструктивный тип нарушения вентиляции | Без особенностей | При малых пороках без особенностей, при больших — рестриктивный тип нарушения |
|
Бактериологическое исследование мокроты |
Стафилококковая, гемофильная, синегнойная инфекции с раннего возраста | Без особенностей | Без особенностей | Чаще пневмококк, м.б. микробные ассоциации, госпитальные штаммы |
| Прогноз | Тяжелый, часто погибают в детском возрасте от дыхательной недостаточности, поражения печени, инфекционных осложнений. | Благоприятный | Благоприятный | Благоприятный |
Диагноз МВ подтверждается при наличии одного или более фенотипических проявлений МВ в сочетании с доказательствами наличия мутации гена МВТР: клинически значимые мутаций гена МВТР при генотипировании или увеличение уровня хлоридов в секрете потовых желез больного.
Для подтверждения диагноза достаточно иметь два признака, по одному из каждого столбца (табл. 5).
Таблица 5 – Диагностические критерии муковисцидоза [9]
| Характерные клинические проявления МВ (патология органов дыхания,придаточных пазух носа, желудочно-кишечные нарушения, нарушения питания, синдром потери солей, обструктивная азооспермия) | Плюс | Положительный потовый тест |
| МВ у сибсов | Две значимые мутации в гене МВТР |
Другим вариантом диагностических критериев является комбинация признаков (табл.6) [17]
Таблица 6 — Диагностические критерии:
|
Положительная потовая проба и/или Две мутации МВТР, вызывающие МВ (согласно базе CFTR-2) |
| И |
|
Неонатальная гипертрипсиногенемия или Характерные клинические проявления, такие как диффузные бронхоэктазы, высев из мокроты значимой для МВ патогенной микрофлоры (особенно синегнойной палочки), экзокринная панкреатическая недостаточность, синдром потери солей, обструктивная азооспермия |
Лечение
Препараты (действующие вещества), применяющиеся при лечении
| Азитромицин (Azithromycin) |
| Амброксол (Ambroxol) |
| Амикацин (Amikacin) |
| Амоксициллин (Amoxicillin) |
| Амфотерицин B (Amphotericin B) |
| Ацетилцистеин (Acetylcysteine) |
| Будесонид (Budesonide) |
| Вориконазол (Voriconazole) |
| Гентамицин (Gentamicin) |
| Джозамицин (Josamycin) |
| Дорназа альфа (Dornase alfa) |
| Имипенем (Imipenem) |
| Ипратропия бромид (Ipratropium bromide) |
| Каспофунгин (Caspofungin) |
| Клавулановая кислота (Clavulanic acid) |
| Кларитромицин (Clarithromycin) |
| Колекальциферол (Kolekaltsiferol) |
| Колистиметат натрия (Colistimethate sodium) |
| Меропенем (Meropenem) |
| Микафунгин (Micafungin) |
| Натрия хлорид (Sodium chloride) |
| Офлоксацин (Ofloxacin) |
| Панкреатин (Pancreatin) |
| Пиперациллин (Piperacillin) |
| Преднизолон (Prednisolone) |
| Ретинол (Retinol) |
| Сальбутамол (Salbutamol) |
| Сульбактам (Sulbactam) |
| Сульфаметоксазол (Sulphamethoxazole) |
| Тазобактам (Tazobactam) |
| Тобрамицин (Tobramycin) |
| Токоферол (Tocopherol) |
| Триметоприм (Trimethoprim) |
| Урсодезоксихолевая кислота (Ursodeoxycholic acid) |
| Флуконазол (Fluconazole) |
| Цефаклора (Cefaclor) |
| Цефепим (Cefepime) |
| Цефиксим (Cefixime) |
| Цефоперазон (Cefoperazone) |
| Цефтазидим (Ceftazidime) |
| Цефтриаксон (Ceftriaxone) |
| Цефуроксим (Cefuroxime) |
| Циластатин (Cilastatin) |
| Ципрофлоксацин (Ciprofloxacin) |
| Эргокальциферол (Ergocalciferol) |
Лечение (амбулатория)
ТАКТИКА ЛЕЧЕНИЯ НА АМБУЛАТОРНОМ УРОВНЕ [1-9, 14,17, 21, 42-45,46, 51,53,58,59]
Цели лечения:
- Обеспечить максимально высокое качество жизни пациента;
- Предупреждение и лечение обострений хронического инфекционно-воспалительного процесса в бронхолегочной системе;
- Обеспечить адекватный рацион и режим питания.
Обязательные составляющие лечения:
- Методики дренирования бронхиального дерева и лечебная физкультура;
- Диетотерапия;
- Муколитическая терапия;
- Бронхолитическая терапия;
- Антибактериальная терапия;
- Заместительная терапия недостаточности экзокринной функции поджелудочной железы;
- Противовоспалительная терапия;
- Витаминотерапия.
Немедикаментозное лечение [1, 21, 42-45]
Диетотерапия — важная часть комплексной терапии при МВ [1, 21, 42-45]. Нарушения нутритивного статуса при МВ являются следствием мальабсорбции вследствие хронической панкреатической недостаточности; недостаточной эмульгации жиров вследствие билиарной недостаточности, снижения активности панкреатических и кишечных ферментов. Диета при МВ должна быть приближена к нормальной, богатой белками, калориями, без ограничений в количестве жиров (УД – B). Необходима ранняя «агрессивная» нутритивная терапия. Существует прямая корреляция между показателем индекса массы тела, функцией легких и продолжительностью жизни. Необходимо повышенное количество белка, источники — натуральные продукты (мясо, птица, рыба, морепродукты, молоко, кисломолочные продукты, творог, сыры, яйца). Детям старше года рекомендуется включение высокобелковых продуктов (яйца, рыба, творог, сыр) не реже 3 раз в день, молоко и кисломолочные продукты не менее 500–800 мл/день. Всем пациентам при наличии панкреатической недостаточности рекомендован дополнительный источник белка — лечебные смеси для энтерального питания по 150 — 250 мл 1-3 раза в день (как второй завтрак, полдник, перед сном). Объем дополнительного питания определяется степенью нутритивной недостаточности.
Важно высокое потребление жиров, которые являются энергетически «плотным» энергоносителем (9 ккал/г); путем адекватной заместительной ферментной терапии. Важен качественный состав жиров: количество насыщенных и транс-жиров ограничивается; предпочтительны полиненасыщенные жирные кислоты (ПНЖК) омега-3, содержащиеся в растительных маслах, жире морских рыб. Энергетическую плотность увеличивают дополнительным питанием смесями с включением среднецепочечных триглицеридов (СЦТ) с 40-70% жирового компонента, в т.ч. специальные препараты СЦТ.
Энергетический дефицит восполняется также углеводами. Простые углеводы не ограничиваются, однако, учитывая риск ассоциированного с МВ сахарного диабета, их рекомендуется употреблять после основных приемов пищи. Ограничение лактозы не требуется. Мальтодекстрины имеют более низкую осмолярность, чем моно- и дисахариды, их использование в составе смесей увеличивает калорийность без увеличения осмотической нагрузки на кишечник. Амилорея, креаторея, стеаторея корректируются панкреатическими заменителями.
Соль не ограничивается: с грудного возраста блюда прикорма подсаливают, у более старших детей и взрослых — подсаливание пищи по вкусу.
При диабете, связанном с МВ: калорийность рациона и содержание жиров остается повышенным! Коррекция уровня сахара крови — за счет инсулина.
Потребность в энергии при МВ должна быть повышена до 150-200% по сравнению с расчетами на фактический вес. Калорийность суточного рациона рассчитывается на долженствующий вес [21, 42-45] (Ур.док. B). Энергетическая потребность обеспечивается жирами на 35-45%, 15% белками и 45-50% — углеводами. Белок дается в расчете 200% от возрастных нормативов.
Используются следующие рекомендации по дополнительному питанию: 1-2 года — 200 ккал, 3-5 лет — 400 ккал, 6-11 лет — 600 ккал, старше 12 лет — 800 ккал в сутки [21, 42-45].
Таблица 7 — Рекомендуемые величины потребления белка и энергии:
| Возраст | Белок, г/кг/сут | Энергия, ккал/кг/сут | |
| Минимальная | Максимальная | ||
| 0 — 1 год | 3 — 4 (до 6) |
|
|
| 1 — 3 года | 4 — 3 | 90 — 100 |
|
| 3 — 10 лет | 3 – 2,5 | 70 — 80 |
|
| 11-14 лет | 2,5 — 1,5 | 45 — 70 |
|
Недостаточность питания констатируется, если процент соответствия массы по росту и полу или массо-ростовой индекс (МРИ), фактическая масса/идеальная масса по росту и полу х 100% меньше 90% у пациентов детского возраста, а у подростков и взрослых — если ИМТ менее 18,5 кг/м2. Ведение пациентов с МВ осуществляют в зависимости от их физического статуса (табл.8). При показаниях Z-score по ИМТ от -1 до -2 диагностируют среднетяжелую недостаточность питания, при> -2 – тяжелую недостаточность питания (УД – B) [21, 42-45].
Таблица 8 — Диетологические рекомендации по ведению пациентов с МВ в зависимости от физического статуса:
| Возраст | <2 лет | 2 – 18 лет |
| Нормальное состояние питания — профилактическое консультирование | МРИ = 90-110% | МРИ = 90-110% |
| После пересмотра режима питания рассмотреть необходимость введения специальных смесей | Любое снижение темпов увеличения массы тела | МРИ = 85-89% или потеря массы тела в последние 4-6 месяцев наблюдения или отсутствие его нарастания через 6 месяцев наблюдения |
| Агрессивное питание: через гастростому и назогастральный зонд; парентеральное питание | Невозможность улучшить нутритивный статус на фоне применения дополнительного энтерального питания | МРИ или падение массы тела ниже 2 перцентиля на фоне применении дополнительного энтерального питания |
Дети первого года жизни. У детей первых месяцев жизни оптимальной пищей является материнское молоко с добавлением микрогранулированных панкреатических ферментов в каждое кормление [21, 42-45, 51, 59-61, 90] (УД – В). Идеальным является непастеризованное грудное молоко, что ассоциируется с лучшими показателями легочных функций, низкой частотой инфекционных эпизодов. При невозможности новорожденным самостоятельно высасывать необходимый объем молока ввиду тяжести состояния, получают сцеженное непастеризованное материнское молоко из бутылочки или через назогастральный зонд. При недостаточной прибавке в весе молоко обогащают добавлением на каждые 100 мл 5 г сухой смеси на основе гидролизата белка со среднецепочечными триглицеридами (СЦТ).
При смешанном/искусственном вскармливании рекомендуются высококалорийные смеси, содержащие СЦТ и растительные жиры в эмульгированной форме. У детей, сохраняющих удовлетворительные темпы физического развития, могут использоваться обычные адаптированные молочные смеси. Для детей с МВ не рекомендуется использовать заменители с низким (1,1 – 1,3 г/100 мл) содержанием белка. При гипотрофии назначают смеси на основе гидролизатов белка с включением СЦТ не менее 50% жирового компонента (УД – В).
При повторных курсах антибактериальной терапии рекомендовано ввести адаптированную кисломолочную смесь или смесь, обогащенную пробиотиками, в количестве до 1/3 суточного объема.
Прикорм вводится в 4–5 мес., при низкой прибавке в массе раньше (табл. 9) [21, 32, 33, 42-45, 51, 59-61] (УД – В). Первыми блюдами прикорма служат энергетически плотные блюда: каши на сцеженном молоке/молочной смеси со сливочным маслом, творог 4,5-5% жирности, овощное пюре с мясным пюре и растительным маслом, желток. Используют высококалорийные продукты прикорма, обогащенные витаминно-минеральным комплексом: детские молочные каши промышленного производства с добавлением сливочного масла, овощные пюре с добавлением растительного масла, мяса. Возможно введение мясного пюре в качестве первого прикорма. Коровье и козье молоко можно использовать с 8–9 мес. В эти же сроки вводят неадаптированные кисломолочные продукты (кефир, натуральный йогурт), обогащенные живыми бифидо- и лактобактериями. Детям раннего возраста назначают поливитаминные добавки; блюда прикорма подсаливают. Дополнительное количество поваренной соли в день составляет 1/8 чайной ложки (0,6 – 0,7 г) в первом полугодии и ¼ ч. л. (1,25 г) для ребенка 6-12 месяцев.
Таблица 9. Особенности введения прикорма детям первого года жизни с МВ:
| Продукты и блюда | Возраст (мес.) |
| Фруктовое пюре | 6 |
| Творог | 4 – 4,5 |
| Желток | 5 |
| Пюре овощное | 4,5 – 5 |
| Масло растительное | 4,5 – 5 |
| Каша | 4 (на грудном молоке, молочной смеси или гидролизате белка) |
| Масло сливочное | 4 |
| Пюре мясное | 5 – 5,5 |
| Молоко | 8 – 9 (для приготовления блюд) |
| Кефир, йогурт | 8 – 9 |
| Сухари, хлеб | 7 – 8 (пшеничный, высшего сорта) |
Питание дошкольников, школьников и взрослых, больных МВ [1, 2, 9, 12, 21, 32, 33, 42-45, 51, 59-61].
Основной принцип – активный подход к питанию в любом возрасте. Питание должно быть регулярным (6 раз/день, формула 3+3): 3 основных (завтрак, обед, ужин) и 3 дополнительных перекуса (2-й завтрак, полдник, на ночь).
Питание должно быть «плотным»: в каждый основной прием пищи должны включаться блюда, содержащие качественные животные белки, цинк (мясо, рыба, яйца, молочные продукты), качественные жиры (растительное масло – рапсовое, соевое, льняное, тыквенное, оливковое, в меньшей степени – подсолнечное, кукурузное, сливочное масло, сметана, сливки), сложные (крупы, хлеб, овощи) и, в меньшей степени, простые (сладости, варенье, мед) углеводы. Дополнительные приемы пищи (2-й завтрак, полдник, перед сном) обязательны; состоят из кисломолочных продуктов, творога, фруктов, выпечки, умеренного количества сладостей.
При бронхолегочных обострениях, значительном отставании в весе для перекусов используют специализированные высокоэнергетические коктейли или смеси для энтерального питания.
Показанием к дополнительному питанию является любое снижение нормальных (возрастных) прибавок массы тела/роста; фактическая масса тела ниже 25-го перцентиля.
К высококалорийным относятся смеси, содержащие более 70 ккал/100 мл для детей до 12 мес, от 100 до 150 ккал – для детей 1-6 лет; от 150 до 200 ккал – для детей старше 7 лет и взрослых. Для дополнительного питания подростков и взрослых с МВ не рекомендуется использование специальных продуктов и смесей для спортивного питания.
Диетологическая профилактика ассоциированной с МВ болезни печени и ассоциированного с МВ сахарного диабета [21, 32, 33, 42-45, 51, 59-61]. При МВ до 10% к подростковому возрасту имеют цирроз печени; до 50% к 30-летнему возрасту сахарным диабетом (CFRD). Нежелательны продукты:
- увеличивающие нагрузку на печень, желчевыводящие пути, поджелудочную железу: транс-жиры (жареные блюда, копчености, колбасные изделия промышленного производства, мясные деликатесы, кулинарный жир, маргарин, в т.ч. в составе продуктов — выпечки, печенья, кондитерских изделий);
- содержащие большое количество стабилизаторов, искусственных красителей, консервантов: майонез промышленного производства, фастфуд, так называемая «мусорная пища» – junk food: чипсы, сухарики, лапша мгновенного приготовления, готовые сухие полуфабрикаты, сладкие газированные напитки: лимонады кока-кола, фанта, спрайт, неразбавленные сладкие фруктовые напитки («нектары») промышленного производства в большом количестве и отдельно от других приемов пищи — рафинированные простые углеводы (сахар, конфеты-леденцы);
- при сохраняющихся диспептических явлениях (увеличение живота, боли в животе, метеоризм, частый стул, выпадение прямой кишки) — большие объемы продуктов, усиливающих газообразование в кишечнике: цельнозерновой и отрубной хлеб, свежая и кислая белокочанная, краснокочанная капуста, бобовые, свекла, кожица и семечки от фруктов, орехи, грибы, неразбавленные соки;
- при формировании сахарного диабета, ассоциированного с муковисцидозом (CFRD), калорийность рациона и содержание жиров (в отличие от диабета 1-го и 2-го типа) сохраняются повышенными.
При МВ важно дополнительное подсаливание пищи, обогащение ее длинноцепочечными полиненасыщенными жирными кислотами (ДЦПНЖК), кальцием.
Для больных с МВ важно адекватное наличие микроэлементов и витаминов [21, 32, 33, 42-45, 51, 59-61, 90].
Таблица 10 — Базовая потребность в электролитах.
| Возрастная группа | Na (ммоль/кг) | K (ммоль/кг) | Cl (ммоль/кг) |
| Новорожденные | 2–3 | 1,5–3 | 2–3 |
| Дети до года | 2–3 | 2–3 | 2–4 |
| Дети младшего возраста | 2–3 | 1–2 | 2–3 |
| Школьники-взрослые | 1–3 | 1–2 | 2 |
Суточная потребность в натрии складывается из физиологической потребности и дефицита, вызванного патологическим процессом.
Пациентам с МВ рекомендовано дополнительное введение кальция: 400-800 мг детям; 800-1200 мг подросткам и взрослым.
Витаминотерапия
Рекомендовано назначение жирорастворимых витаминов, низкие уровни которых регистрируются практически у всех больных. Умеренно повышенные уровни ретинола (до 110 мг/дл) в сыворотке крови положительно коррелируют с ОФВ1 (ОФВ1>80%). У нелеченых больных МВ геморрагический синдром может манифестировать витамин-К недостаточностью. У новорожденных и грудных детей она может проявиться необъяснимой геморрагической пурпурой, интестинальными кровотечениями, длительной кровоточивостью в местах инъекций. При интенсивной антибактериальной терапии, при поражении печени нарушены процессы коагуляции. При МВ недостаточно витамина Д, нарушен фосфорно-кальциевый обмен; все больные МВ с панкреатической недостаточностью ежедневно должны получать дополнительно жирорастворимые витамины (А, Д, Е и К) и бета-каротин; с сохранной функцией поджелудочной железы — витамин Е.
Водорастворимые витамины назначаются больным МВ в обычных дозировках, за исключением витамина С, потребность в котором у больных повышена, и витамина В12 случаях резекции подвздошной кишки.
Таблица 11 — Рекомендуемые дозы жирорастворимых витаминов и бета-каротина для пациентов с МВ.
| Витамины | Характеристика больных | Дозы |
| А | Все с ПН* | 4000-10000 МЕ/сут** |
| Д | Все с ПН* | 400 — 2000 МЕ/сут** |
| Е |
Все: 0-6 мес 6-12 мес 1-4 года 4-10 лет Старше 10 лет |
25 МЕ/сут ** 50 МЕ/сут 100 МЕ/сут 100-200 МЕ/сут 200 – 400 МЕ/сут |
| К |
Все с ПН при патологии печени |
2– 5 мг/сут 2-10 мг/сут |
| Бета-каротин | Все с ПН | 0,5 – 1 мг/кг/сут, макс. 50 мг/сут |
| *ПН – панкреатическая недостаточность | ||
|
**Перевод одних единиц измерений доз витаминов в другие: витамин А: 1 мг = 3333,3 МЕ витамин Д: 1 мкг = 40 МЕ; витамин Е: 1 мг = 1,36 МЕ |
Агрессивные методы нутритивной поддержки у пациентов с МВ [21, 42-45, 59-61, 90]:
Показания к применению «агрессивных» методов нутритивной поддержки:
У детей:
- отсутствие прибавки в весе или снижение веса в течение 6 месяцев;
- фактическая масса тела ниже 3 перцентиля (или Z-score ИМТ/возраст, масса тела/возраст, масса тела/рост менее -2);
- фактическая масса тела ниже должного значения на 15% или менее 25 перцентиля на фоне дополнительного питания специальными смесями.
У взрослых:
- ИМТ <18,5 или снижение массы тела более чем на 5% за период менее 2 месяцев;
- Невозможность улучшить нутритивный статус на фоне дополнительного приема высококалорийных смесей.
«Агрессивные» методы нутритивной поддержки при МВ:
1. Зондовое энтеральное питание в виде ночной гипералиментации, через назогастральный зонд или через перкутанную гастростому [21]. Используют смеси для энтерального питания, вводят капельно (с помощью инфузионного насоса) в ночное время в течение 5-6 часов. Могут использоваться как полуэлементные смеси (на основе гидролизатов белка), так и полимерные (последние предпочтительны ввиду относительно низкой осмолярности). Ночную гипералиментацию начинают с 1/3 рассчитанной от суточной потребности в калориях и увеличивают по мере прибавки в весе. Объем смеси для ночной гипералиментации подбирается так, чтобы не снижался аппетит в дневное время. Заместительная ферментная терапия проводится минимикросферическими препаратами панкреатина. Оптимальна установка низкопрофильной гастростомы, которая позволяет поддерживать активный образ жизни, не мешает спортивным занятиям и проведению кинезитерапии.
2. Парентеральное питание:
- Полное (центральный венозный катетер): при состояниях после операции на кишечнике; синдроме короткой кишки; остром панкреатите.
- Частичное (с целью дополнительного питания), используется периферическая вена: жировые эмульсии; глюкозо-аминокислотные смеси, витамины.
Таблица 12 — Виды питательных смесей при МВ:
| Наименование |
|
|
|
|
Для дополнительного энтерального питания рекомендуются применение питательных смесей с учетом возраста, суточных потребностей, отклонений в нутритивном статусе.
Разновидности питательных смесей для энтерального питания:
- на основе цельных белков молока или частичного гидролиза белка;
- на основе белков молока;
- на основе глубоких гидролизатов белка, с включением СЦТ в состав жирового компонента;
- молочные коктейли и высококалорийные пудинги;
- масла, содержащие среднецепочечные триглицериды.
Режим:
Очень важно соблюдение санитарно-гигиенического режима при МВ (проветривание помещений, исключение контакта с инфекционными больными, чистое нательное и постельное белье и др.).
Общие меры профилактики: обязательно использование соответствующих мер профилактики инфекций как у пациентов, так и медработников при работе с пациентами МВ. Контакты между пациентами МВ должны быть сведены к минимуму.
Кинезитерапия (лечение с помощью движения) [2, 6, 9, 13-15, 17, 18].
Принципы кинезитерапии:
- от простого упражнения к сложному;
- адаптировать методики для пациента, а не наоборот;
- отсутствие резких движений;
- занятие должно приносить пациенту облегчение и удовольствие.
Кинезитерапия включает физические упражнения, специальные дыхательные маневры и положения, вибрационный массаж грудной клетки, аппаратная гимнастика, аппаратную дренажную вибротерапии грудной клетки, обучение технике ингаляции.
— Дренажные положения:
Режим дозирования: 1 и/или 2 раза в день, до еды/через 1 час после еды и за 2 часа до сна, по 15-20 мин; во время одной дренажной позиции делается 6-7 вдохов; чередуясь друг с другом (на 1 занятие приходится не более 3-х различных положений); в лечебный комплекс каждое новое упражнение вводится постепенно, не более 1-го нового положения, обучаясь 5-6 дней.
— Клопфмассаж с использованием 8-12 точек в последовательных дренажных положениях [51].
— Аутогенный дренаж (АДР) — для пациентов более осознанного возраста – с соблюдением соответсвующих фаз и дыхательных маневров
— Ассистированный аутогенный дренаж (ААДР). основан на принципах аутогенного дренажа и используется у пациентов, которые неспособны проводить АДР самостоятельно [58].
— Применение специальных дыхательных тренажеров PEP (Positive Expiratory Pressure, положительное давление на выдохе) системами (с созданием положительного давления в условиях ЛПУ и/или самостоятельной PEP-терапии; для вибрационной PEP-терапии; нагрузочные/побудительные спирометры, работающие по объему и потоку; флаттеры для облегчения откашливания и др.) [13-15, 53, 59].
— Аппаратные методы кинезитерапии, особенно у детей младшего возраста и тяжелых пациентов с помощью вибрационного воздействия [13-15, 51]: в виде вибро-жилетов различных размеров, в зависимости от возраста и размера грудной клетки пациента; виброакустическая терапия (ВАТ): сочетание вибрационного и акустического воздействия волнами высокой интенсивности [52].
Дыхательные упражнения [2, 6, 9, 13-15, 17, 18]: наряду с кинезитерапией применяются дыхательные упражнения:
1. «БАНАН» положение на спине. Руки вытянуты вверх и направлены в одну сторону (влево или вправо). Туловище максимально изогнуто. Ноги вытянуты прямо и направлены в туже сторону, что и руки.
2. «ВИНТ» Голова и верхняя часть туловища прижаты к полу, касаясь его лопатками. Руки вытянуты вверх. Нижняя часть туловища повернута набок в одну сторону (влево или вправо). Нижняя нога вытянута. Верхняя нога максимально согнута в колене.
3. «КОБРА» положение на животе. Ноги вытянуты. Руки тянутся назад до ягодиц. Затем, голова и верхняя часть туловища поднимаются вверх.
4. «УЗЕЛ» Положение сидя. Правая нога согнута в колене, направлена влево и помещена за левой коленкой. Верхняя часть туловища повернута вправо. Правая рука, в виде упора, позади ягодиц, как можно дальше влево. Кончики пальцев направлены назад. Локоть левой руки прижат к правой коленке. Затем в другую сторону.
5. «КУВЫРОК» Из положения сидя вращаться на спине назад, до тех пор, пока колени не коснутся ушей. Руки поддерживают ягодицы.
6. «ГОРКА» положение сидя на пятках. Руки назад, как упор. Варианты упражнения:
— кончики пальцев направлены назад. Грудь приподнять вверх.
— из положения «сидя на пятках» медленно двигаться вперед с вытянутыми руками. Ягодицы позади коленей.
7. «ЖИРАФ» Стоя на коленях, левая рука вытянута вверх. Туловище поворачивается вправо вслед за правой рукой, которая должна коснуться левой пятки. Затем, в другую сторону
8. «ПТИЧКА» В положении лежа на животе верхнюю часть туловища и голову приподнять. Одна рука вытянута вперед. Другая рука сжимает стопу противоположной стороны.
Для детей 2-5 лет удобно используют упражнения: «банан», «винт», «кувырок», «горка», «жираф», «птичка».
Для более старших и взрослых: «кобра», «узел», «винт», «рыбка», «птичка», «кувырок».
Таблица 14 — Виды спорта, разрешенные и запрещенные у больных муковисцидозом.
| Разрешено | Запрещено* |
| Плавание | Коньки |
| Бег | Тяжелая атлетика |
| Езда на велосипеде | Футбол |
| Лыжи | Хоккей |
| Бадмингтон | Бокс |
| Большой и малый теннис | Прыжки в воду |
| Верховаяезда | Регби |
| Йога | Дзюдо |
| Ушу | Баскетбол |
| Волейбол | Мотоспорт |
| Гольф | |
| Туризм |
*Примечание: эти виды спорта запрещены в связи с опасностью, повышенной травматизации, следствием которой является длительный период ограничения физической активности, что крайне неблагоприятно сказывается на дренажной функции легких.
Кислородотерапия
Одним из тяжелых осложнений МВ является хроническая дыхательная недостаточность (ХДН). Главным признаком ХДН является гипоксемия (снижение содержания кислорода в артериальной крови). Коррекция гипоксемии проводится длительной кислородотерапией (не менее 15 часов/сутки (УД – В)). Проводится в домашних условиях — длительная кислородотерапия (ДКТ); назначение кислорода (> 15 часов/сутки) увеличивает выживаемость у больных с гипоксемией в покое (УД – В). Используют концентраторы кислорода. ДКТ назначается при стойкой гипоксемии (SpO2<89% и/или PaО2<55 мм рт.ст.) не менее 16 час/сут. При гипоксемии только на фоне физических нагрузок аппарат используется во время нагрузок, при ночной гипоксемии — ночью. Контроль проводится пульсоксиметрией и исследованием газов артериальной крови.
Задача ДКТ — достижение значений РаО2>60 мм рт.ст. и SaO2>90%. Не следует добиваться увеличения показателей пульсоксиметрии выше 94–95%, так как в этом случае возможно развитие гиперкапнии.
Медикаментозное лечение
Среди медикаментозной терапии наибольшее значение имеют:
- Муколитическая терапия;
- Бронхолитическая терапия;
- Заместительная терапия недостаточности экзокринной функции поджелудочной железы;
- Антибактериальная терапия;
- Противовспалительная терапия;
- Витаминотерапия
Муколитическая терапия [9, 17, 46]. Цель — нормализация вязкоэластических свойств секрета и оптимизация мукоцилиарного транспорта. Обязательны достаточная гидратация и кинезитерапия. Применение противокашлевых препаратов противопоказано [9, 46].
Путь введения предпочтительно ингаляционный. Оценка эффективности проводится клинически (оценка суточного дебета мокроты, изменений ее консистенции, оценка степени дыхательной недостаточности).Классы муколитиков при МВ:
— Дорназа альфа — раствор рекомбинантной человеческой дезоксирибонуклеазы; разрывает водородные связи молекул ДНК, действует на все звенья «порочного круга» МВ – обструкцию, инфекцию, воспаление (УД А). Применяется ингаляционно в дозе 2,5 мг 1 раз в сутки (в тяжелых случаях может вводится в два приема). Вводится при помощи джет-небулайзера/компрессора многоразового пользования, или меш-небулайзера. Является базовым муколитиком и назначается всем больным МВ сразу после постановки диагноза [1, 2, 4, 6, 12, 13, 18, 24].
— Гипертонический раствор NaCl 3–7%, ингаляции (повышает концентрацию соли в бронхиальном секрете, приводит к его увлажнению, улучшению мукоцилиарного транспорта (УД B). При необходимости применяют бронходилататоры до гипертонического раствора [1, 2, 6, 12, 13, 47, 48]. Возможна комбинация гипертонического NaCl с 0,1% гиалуроновой кислотой (ГК), что блокирует возникновение бронхоспазма, облегчает вентиляцию и газообмен [1, 2, 6, 12, 13, 49].
– Маннитол в форме порошка для ингаляций (УД B) [1, 2, 6, 12, 13, 50]. Применяется при отсутствии значимого эффекта от лечения дорназой альфа и/или плохой переносимости гипертонического раствора. В дозе 400 мг дважды день через ингалятор. Перед применением рекомендуется предварительное использование бронходилататоров.
— N-ацетилцистеин ингаляционно, внутрь, в/в. Применяется из расчета 30мг/кг/сут в 2-3 приема (УД В) [1, 2, 6, 12, 13].Бронхолитические препараты рекомендованы пациентам с МВ (УД В) [1, 2, 6, 12, 13]:
— при возникновении затрудненного дыхания и одышки;
— перед проведением кинезитерапии;
— перед проведением ингаляционной терапии муколитиков.
β2-адреномиметики в виде монотерапии (сальбутамол), а также комбинированные препараты в сочетании с М-холинолитиком (фенотерол+ипратропия бромид). При необходимости могут применяться пролонгированные β2-адреномиметики (сальметерол, формотерол) (УД В-С) [1, 2, 6, 12, 13].
Заместительная терапия недостаточности экзокринной функции поджелудочной железы (панкреатические ферменты) [1, 2, 6, 12, 13]. Коррекция панкреатической недостаточности при МВ проводится только препаратами панкреатина в микросферической форме (микрогранулы от 0,4 до 2 мм) [1, 59, 60, 61]. Адекватность замещающей панкреатические ферменты терапии определяют клинически, по алиментарному статусу [1, 59, 60].
Дозирование панкреатических ферментов при муковисцидозе. Детям с сохранной функцией поджелудочной железы в момент постановки диагноза назначение препаратов не рекомендуется, если физическое развитие ребенка не страдает. Заместительную панкреатическую терапию назначают сразу после установления диагноза, целевые показатели всасывания жиров составляют от 85 до 95% [1, 17, 19, 42, 45]. Доза панкреатина индивидуальна, подбор начинают в зависимости от массы тела (1000 ЕД/кг по липазе на каждый прием пищи для детей младше 4-х лет и 500 ЕД/кг — старше 4-х лет и взрослых), в дальнейшем доза может постепенно повышаться.
Новорожденным на каждые 120 мл питания стартовая доза рассчитывается как 2500-3333 ЕД липазы (1/4-1/3 капсулы препарата с активностью 10 000 ЕД липазы в капсуле). Дозы соответствуют примерно 600-800 ЕД липазы на 1 г пищевых жиров [1, 17, 19, 42, 45]. Рекомендации по дозированию панкреатических ферментов (Табл. 15) [45].
Таблица 15. Заместительная панкреатическая терапия, рассчитанная по содержанию липазы:
| Возраст | Предлагаемая дозировка |
| Дети грудного возраста (до 12 месяцев) | 2000-4000 ЕД липазы/120 мл грудного молока или молочной смеси, что примерно равно 2000 ЕД липазы на 1 г жира в пище |
| Дети от 1 до 4 лет | 2000-4000 ЕД липазы на 1 г жира в пище, повышая по необходимости (максимальная дозировка – 10 000 ЕД липазы/кг массы тела в сутки) |
| Дети старше 4 лет и взрослые | Начиная с 500 ЕД липазы/кг массы тела на прием пищи, повышая постепенно до максимальной дозы, которая составляет: — 1000-2500 ЕД липазы/кг массы тела на один прием пищи, или — 10 000 ЕД липазы/кг массы тела в сутки, или — 2000-4000 ЕД липазы на 1 г жира со всеми содержащими жиры приемами пищи, перекусами, напитками |
Ферментная терапия при энтеральном питании назначается с учетом степени экзокринной недостаточности.
- Доза фермента подбирается из расчета: от 500 ЕД липазы (дети с относительной панкреатической недостаточностью) до 4000 ЕД липазы (абсолютная панкреатическая недостаточность) на 1 г жира в пище.
- Рекомендуется корректировка в соответствии с кишечными симптомами, стеатореей и увеличением веса.
- На среднецепочечные триглицериды (СЦТ) дополнительного назначения ферментов не требуется. Гидролизованные и элементные смеси требуют назначения ферментов из расчета 2000-4000 ЕД липазы на 1 г жира.
Антибактериальная терапия [1–3, 6, 9, 12, 15-19, 33, 36-38, 46, 51, 62-68] (АБТ) респираторной инфекции определяет прогноз заболевания. При МВ в отмечается носительство Staphylococcus aureus, Haemophilus influenzae, Streptococcus pneumoniae, Pseudomonas aeruginosa, Burkholderia cepacia complex. В первые годы жизни доминируют St.aureus и Haemophilus influenzae, затем – Ps.aeruginosa. В 2/3 случаев хроническая инфекция вызвана ассоциацией микроорганизмов. Высока роль Nontuberculous mycobacteria, неферментирующих грамотрицательных микроорганизмов Stenotrophomonas maltophilia, Аchromobacter (Alcaligenes) spp., Аspergillus spp. Продолжительность жизни больных МВ зависит от этиологии хронической легочной инфекции.
Показания для назначения антибиотиков при МВ:
- с профилактической целью;
- для первичной эрадикации возбудителя;
- для лечения хронической легочной инфекции;
- для лечения обострений легочной инфекции.
Особенности АБТ при МВ [1–3, 16-19, 46, 62-65]
- При проведении АБТ ориентируются на чувствительность выделенного микроорганизма/микробной ассоциации, а также на положительный эффект при терапии предшествующего обострения. При выделении 2-х микроорганизмов выбор АБП зависит от свойств более резистентного возбудителя. Резистентность к антибактериальным препаратам (АБП) не является основанием для изменения лечения в случае положительного ответа на проводимую терапию.
- Назначаются максимальные возрастные дозы препарата.
- При обострении легочной инфекции предпочтительно использовать внутривенный путь доставки, который начинается в стационаре и продолжается в амбулаторных условиях.
- Одновременное назначение препаратов для ингаляционного и внутривенного путей введения одной фармакологической группы не рекомендуется.
- Рекомендуется использовать комбинации АБП с различным механизмом действия (например, β-лактамы с аминогликозидами).
- Для базисной терапии хронического микробно-воспалительного поражения легких используется ингаляционная АБТ; при недостаточной эффективности дополняется внутривенным введением или приемом внутрь.
- АБТ необходимо сочетать с активной кинезитерапией.
- Длительность АБТ определяется на основании клинических, лабораторных, рентгенологических и функциональных признаков обострения. Курс лечения составляет от 14–21 день и более.
- Применение АБП в виде ингаляций через небулазер проводится в стационаре и в домашних условиях.
- Перед ингаляцией АБП за 15-30 мин необходимо провести ингаляцию бронхолитиков, муколитиков, а также постуральный дренаж.
- При ингаляции АБП в домашних условиях необходимо использовать небулайзер, оснащенный фильтром для выдыхаемого воздуха.
- При проведении ингаляций в условиях ЛПУ, больные, инфицированные P.aeruginosa и B.cepacia, должны пользоваться индивидуальным небулайзером.
- Применение АБП внутрь используется для терапии обострений МВ в домашних условиях или для профилактического лечения больных, инфицированных P.Aeruginosa.
Применение АБП внутривенно показано:
- при тяжелых обострениях МВ (лечение в условиях ЛПУ);
- для профилактического лечения при инфицировании P.aeruginosa, когда применение АБП внутрь неэффективно (внутривенная терапия в домашних условиях);
- при ухудшении течения заболевания и появлении новых симптомов у больных, получающих АБП внутрь (внутривенная терапия в домашних условиях).
Профилактическое назначение антибиотиков при МВ:
- Основано на данных о быстром возникновении Pseudomonas aeruginosa-инфекции на фоне текущего бактериального или вирусного процесса.
- При острых респираторных инфекциях у пациентов с МВ и стафилококковой инфекцией назначаются амоксициллин, амоксициллин/клавуланат, цефалоспорины 2–3-го пок., азитромицин.
- При хронической синегнойной инфекции при возникновении ОРВИ рекомендуется ципрофлоксацин.
- Рекомендуется проведение профилактических курсов антибактериальной терапии при хронической колонизации нижних дыхательных путей P.аeruginosa. Профилактический прием АБТ практически не сказывается на устойчивости штаммов микроорганизмов, но только при своевременной смене препаратов. При частых обострениях инфекционно-воспалительного процесса следует увеличивать продолжительность курсов АБТ до 3 нед, используя внутривенный способ введения, и (или) сокращать интервалы между курсами, и (или) между курсами принимать внутрь ципрофлоксацин [17-19,46,62-65].
Фармакокинетика антибиотиков и особенности их проникновения в респираторный секрет при МВ.
Антибиотики проникают в бронхиальный секрет из крови путем диффузии. Аминогликозиды (АМГ) плохо диффундируют в просвет бронхов [15, 16].
Тобрамицин проникает в бронхиальный секрет лучше других АМГ, для достижения терапевтической концентрации при внутривенном введении необходимы высокие дозы. Антибиотики этой группы обладают длительным (3–4 час) постантибиотическим эффектом, что послужило основанием режима их дозирования — одноразовое введение суточной дозы. АМГ применяются в качестве базисной ингаляционной терапии (УД А) [18–20]. Пенициллины и цефалоспорины плохо проникают в бронхиальный секрет, их концентрация в мокроте составляет 3–15%. При таблетированном применении ципрофлоксацина его концентрация в бронхиальном секрете достигает 46–90% [1,2, 16, 17, 19, 38, 46].
Колистиметат натрия применяется для ингаляционной терапии (с обязательной предшествующей ингаляцией бронхолитиков, муколитической и кинезитерапией) при первом высеве и при хронической P.aeruginosa-инфекции. Внутривенно Колистиметат натрия применяется при инфекции, вызванной мультирезистентной P.aeruginosa. С противовоспалительной целью при P.aeruginosa-инфекции используют макролиды (Ур.доказ.В) [66-69].
Микробиологический статус пациента с МВ:
Выделяют 4 группы больных по результатам бактериологического исследования микрофлоры дыхательных путей за последние 12 мес [40]:
- с хронической синегнойной инфекцией: пациенты, у которых P.aeruginosa идентифицировалась более чем в 50% образцов мокроты или фарингеальных смывах в течение предшествующих 12 мес;
- с интермиттирующим высевом P.aeruginosa в случае синегнойной палочки менее чем из 50% биообразцов в течение предшествующих 12 месяцев;
- свободные от P.aeruginosa, при отсутствии высева в течение 12-ти последних месяцев;
- никогда не были инфицированы P.aeruginosa.
В клинической практике выделяют пациентов с первым высевом P.aeruginosa. Необходимым условием для применения этих критериев является регулярный, не реже одного раза в 3 мес, бактериологический контроль микрофлоры дыхательных путей. Микробиологический статус определяет выбор АБП, показания для комбинированной терапии, путь введения, продолжительность терапии, режим бактериологического контроля [2, 6, 9, 13-15, 17, 18]
Таблица 16 — Антибиотики, применяемые при высеве из мокроты Staphylococcus aureus и Haemophilus influenzae [1,2,6,9,14,17,18].
| Антибиотик | Доза в сутки для детей | Путь введения | Кратность приема/день |
|
Амоксициллин+ Клавулановая кислота (расчет по амоксициллину) |
50-100 мгкг в сутки | Внутрь | 3-4 |
Азитромицин |
>6мес-10мгкг в день 15-25кг-200мг 26-35кг-300мг 36-45кг-400мг |
внутрь |
1 раз 7-10 дней |
Цефаклор |
До 1 года 125мг 3 раза 1-7лет 250мг 3 раза >7лет 500мг 3 раза |
внутрь | 3 раза |
| Цефиксим |
8мг/кг 6мес-1год 75мг 1-4года-100мг 5-10лет-200мг 11-12лет-300мг |
внутрь | 1-2 раза |
| Сульфаметоксазол и Триметоприм |
6н.-5мес 120мг 2 раза 6мес-5лет-240мг 2раза 6-12лет-480мг 2раза при тяжелой инфекции возможно увеличение дозы на 50% |
Внутрь | 2 раза |
| Кларитромицин |
15мг/кг 1-2 г-125 мг 3-6 лет-250 мг 7-9 лет-375 мг >10 лет-500 мг |
внутрь | 2 раза |
| Цефоперазон +Сульбактам |
Дети от 1 мес до 12 лет -80 мг/кг по цефоперазону Для тяжелых инфекций до 160 мг/кг |
в/венно | 2 раза |
| Сульфаметоксазол и Триметоприм |
6-10 мгкг по триметоприму До 5мес 240 мг 6 мес-5лет-480 мг 6-12лет-480 –960 мг Старше 12 лет -1920 мг при тяжелой инфекции возможно увеличение дозы на 50% |
Внутрь | 2-3раза |
| Цефтриаксон | 50-80 мг/кг | в/в, в/м | 1-2раза |
|
Цефуроксим Цефуроксим натрия |
20 -30 мг 150- 200мг/кг |
Внутрь в/венно |
2раза 3-4раза |
Антибактериальная терапия при выявлении в мокроте P.аeruginosa.
Одновременно назначают 2–3 противомикробных препарата из разных групп. Оптимальны комбинации аминогликозидов с цефалоспоринами 3–4 поколения. Целесообразно менять комбинации антибиотиков, лабораторное определение чувствительности не всегда полностью совпадает с клиническим ответом на проводимую терапию [1,2,6,9,14,17,18].
Стратегия антибиотикотерапии P.aeruginosa инфекции при МВ
Активная антимикробная терапия позволяет предупредить или отсрочить развитие хронической P.Aeruginosa инфекции более чем у 80% больных МВ. Если эрадикация P.Aeruginosa после проведенного курса АБТ не произошла, и у больного развилась хроническая синегнойная инфекция, то назначение ингаляционной противосинегнойной терапии (Тобрамицин) позволяет уменьшить риск обострений и степень выраженности респираторных проявлений [1-18].
Таблица 17. Антибиотики при высеве Ps.aeruginosa [1–3, 6, 9, 12, 15-19, 33, 46, 51, 62-68]:
| Антибиотик | Доза в сутки для детей | Суточные дозы для взрослых | Путь введения |
Число приемов в день |
| Амикацин | 15-20 мг/кг | 700-1000 мг | В/в | 2 |
| Гентамицин | 10 мг/кг | 10 мг/кг | В/в | 1-2 |
| Тобрамицин | 300 мг/5мл | 300 мг/5мл | ингаляции | 2 |
| Тобрамицин | 112 мг (4 капсулы по 28 мг) | 112 мг (4 капсулы по 28 мг) | порошок-ингалятор | 2 |
| Цефтазидим | 150-250 мг/кг | 6-9 г | в/в | 2-3 |
| Цефепим | 100-150 мг/кг | 4-6 г | в/в | 2-3 |
| Пиперациллин+Тазобактам | 100-200 мг/кг | 13,5 г | в/в | 3-4 |
|
Цефоперазон+ Сульбактам |
150-200 мг/кг | 8г | в/в | 2 |
| Меропенем | 60-120 мг/кг | 3-6 г | в/в | 3 |
|
Колистиметат натрия |
50-75 тыс ЕД/кг | 6 млн ЕД | в/в | 3 |
|
Колистиметат натрия |
1-4 млн ЕД | 1-4 млн ЕД | ингаляции | 2 |
|
Имипенем+ Циластатин |
50-100 мг/кг в день по имипенему | 2-4 г | в/в | 3-4 |
| Азтреонам | 150-250 мг/кг | 8г | В/в | 4 |
Тобрамицин 300 мг/5мл для ингаляции, в инструкции имеется противопоказание к применению препарата до 6-летнего возраста. Так как инфицирование синегнойной палочкой чаще всего отмечается в раннем детском возрасте (чаще до 3-5лет) [1]. Тобрамицин рекомендуемая дозировка составляет 112 мг (4 капсулы по 28 мг) дважды в сутки в течение 28 дней, что соответствует, 1 ампуле (300 мг тобрамицина) 2 раза в день в течение 28 дней с 6-летнего возраста. Длительность лечения составляет 14 дней и более. Критерием прекращения АБТ является регресс клинических симптомов обострения бронхолегочного процесса (уменьшение хрипов, улучшение дренажной функции бронхов, санация мокроты).
Схемы эрадикационной антибактериальной терапии при первом высеве Ps.aeruginosa [1–3, 16-19, 46, 62-65]: (выбрать одну из схем в зависимости от лекарственного обеспечения, регулярности наблюдения и обследования пациента)
Таблица 18 — Схемы эрадикационной антибактериальной терапии при первом высеве Ps.aeruginosa
| 1-я схема |
|
Проведение санитарно-эпидемических мероприятий 1. 1-й месяц – ингаляции тобрамицин раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день в течение 28 дней. Посев мокроты – через 7-10 дней после окончания лечения. 2. 2-й месяц – перерыв согласно схеме лечения в случае отрицательного высева. 3. 3-й месяц – ингаляции тобрамицин раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день в течение 28 дней. Посев мокроты – через 7-10 дней после окончания лечения. 4. 4-й месяц – перерыв согласно схеме лечения в случае отрицательного высева. 5. 5-й месяц – ингаляции тобрамицин раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день в течение 28 дней. Посев мокроты – через 7-10 дней после окончания лечения. 6. Отрицательный результат посева мокроты – стоп-терапия. |
В случае положительного высева после первого месяца ингаляций тобрамицином:
1. Ингаляции колистиметата натрия 1-2 млн х 2 раза в день 1 мес. Посев мокроты – через 7-10 дней после окончания лечения.
2. Ингаляции тобрамицин раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день, 28 дней. Посев мокроты – через 7-10 дней после окончания лечения.
3. При сохраняющемся высеве Ps.aeruginosa – внутривенная антибактериальная терапия в течение 14 дней двумя антибактериальными препаратами синергидного по чувствительности действия.
4. Затем продолжение чередования ингаляций колистиметат натрия 1-2 млн х 2-3 раза в день с ингаляциями тобрамицина раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день в течение 3 мес.
5. При сохраняющемся высеве Ps.aeruginosa:
a. ингаляции тобрамицина раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день циклами по 28 дней 6 курсов в год
b. или ингаляции колистиметата натрия 1-2 млн х 2-3 раза в день постоянно
c. или чередование колистиметата натрия 1-2 млн х 2 раза в день с тобрамицином раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день, 6 курсов в год каждого.
| 2-я схема |
|
Проведение санитарно-эпидемических мероприятий 1. 1-й месяц a. Ципрофлоксацин 30-40 мг/кг/сутки в 2 приема 1 месяц b. Колистиметат натрия в ингаляциях 1-2 млн х 2 раза в день 1 месяц c. Посев мокроты через 7-10 дней после окончания лечения. В случае отрицательного высева в группе детей до 3 лет без признаков поражения бронхолегочной системы терапию можно прекратить. Бактериологический контроль проводить ежемесячно до 6 месяцев. 2. 2-3-ий месяц – колистиметат натрия в ингаляциях 1-2 млн х 2 раза в день 2 мес. Ципрофлоксацин 30-40 мг/кг/сут в 2 приема 1 мес. Посев мокроты – через 7-10 дней после окончания лечения. 3. При отрицательном высеве – продолжение ингаляций колистиметата натрия 1-2 млн х 2 раза в день 3 мес. При сохраняющемся отрицательном высеве – стоп-терапия. 4. При сохраняющемся высеве Ps.aeruginosa – внутривенная антибактериальная терапия в течение 14 дней двумя антибактериальными препаратами синергидного по чувствительности флоры действия. 5. После курса внутривенной терапии продолжить чередование ингаляций тобрамицина раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день 28 дней, с колистиметатом натрия 1-2 млн х 2 раза в день, 1 мес в течение 3 мес (2 курса тобрамицина и 1 курс колистиметата натрия). 6. При сохраняющемся высеве Ps.aeruginosa терапия назначается по одному из нижеперечисленных вариантов с учетом чувствительности: I. Ингаляции тобрамицина раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день циклами по 28 дней, 6 курсов в год. II. Ингаляции колистиметата натрия 1-2 млн х 2-3 раза в день постоянно. III. Чередование колистиметата натрия 1-2 млн х 2 раза в день с ингаляциями тобрамицина раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг х 2 раза в день, по 6 курсов в год каждого. Назначается при появлении мукоидной формы P. Aeruginosa и/или увеличении степени обсемененности. |
Таблица 19 — Схема антибактериальной терапии при хронической синегнойной инфекции [1–3, 6, 9, 12, 15-19, 33, 46, 62-68]
| Ингаляции тобрамицина раствор 300 мг х 2 раза в день или тобрамицин порошок 112 мг 2 раза в сутки интермиттирующими курсами 28 дней приема, 28 дней перерыв, всего 6 курсов в год. |
| или |
| раствор Колистиметата натрия 1-4 млн. ЕД/24 час постоянно или чередование колистиметата натрия и тобрамицина в ингаляциях постоянно всем с хронической колонизацией Ps.aeruginosa . |
|
У пациентов с прогрессирующим снижением функции легких и частых обострениях, а также при недостаточном эффекте от ингаляционной антибактериальной терапии основной режим терапии включает: 2-х недельный курс внутривенной антимикробной терапии каждые 3 месяца Препараты для внутривенного введения: Тобрамицин 10-12 мг/кг или Амикацин 20 мг/кг 1раз в день (до достижения в сыворотке крови концентрации 1-2 мкг/мл) + Цефтазидим 150-200 мг/кг в день в/в или + Меропенем 60-120мг/кг/24час в/в или + другой антибиотик (азтреонам или др.), активный против Ps.aeruginosa. |
|
При клинической нестабильности: увеличение продолжительности курсов в/в антибактериальная терапия до 3 нед. и/или сокращение интервалов между курсами, и/или прием Ципрофлоксацина 20-40мг/кг/сутки внутрь между курсами в/в антибактериальной терапии, а также непрерывная ингаляционная антибактериальная терапия тобрамицина и колистиметата натрия курсами по 28 дней чередованием длительностью до 2 лет. |
Антибактериальная терапия при выявлении в мокроте B. сepacia [1–3, 6, 9, 12, 15-19, 33, 46, 51, 62-68]. Рекомендуется при инфицировании Burkholderia cepacia complex немедленное терапевтическое вмешательство вследствие ее высокой вирулентности. (УД В). Инфицирование В.cepacia достоверно ухудшает клиническое состояние и прогноз.
Рекомендации по АБТ (как при первичном высеве, так и для лечения обострения бронхолегочного процесса):
1. Комбинация из трех препаратов. Курс от 3 недель и более.
2. Целесообразна комбинация внутривенного и ингаляционного путей и/или перорального введения антибактериальных препаратов.
3. Наиболее эффективны Цефтазидим, Пиперациллин+Тазобактам, Меропенем, Имипенем, Сульфаметоксазол и Триметоприм и Тетрациклины (доксициклин). Для оптимизации исходов «cepacia syndrome» рекомендуется включение в схему лечения Сульфаметоксазол и Триметоприма. Эффективно применение трехкомпонентной схемы в/в введения Меропенема, Тобрамицина с Цефтазидимом в течение 2-х недель и более (табл.20).
4. Эффективна длительная (3-12 недель) терапия пероральными препаратами. Ко–тримаксозолом и/или Доксициклином и/или Хлорамфениколом (на фоне в/в терапии или после нее). При хронической инфекции Burkholderia cepacia рекомендуется прием таблетированных форм Сульфаметоксазол и Триметоприма.
5. При крайне тяжелом течении допустимо сочетание двух лактамных антибиотиков (в/в и ингаляционно). Для детей старше 12 лет и взрослых рекомендовано ингаляционное применение Тобрамицина, Меропенема и Цефтазидима, предназначенных для в/в использования. (УД С).
Об эрадикации Burkholderia cepacia можно судить через год после последнего высева при условии, как минимум, трех отрицательных бактериологических анализов.
Антибактериальная терапия при высеве из бронхиального секрета Achromobacter xylosoxidans [1–3, 6, 9, 12, 15-19, 33, 36, 38, 46, 51, 62-68]. Achromobacter xylosoxidans мультирезистентен.
Рекомендовано соблюдение следующих правил АБТ:
1. При первом высеве используют внутривенные антибиотики курсом 14-21 день: Колистиметат натрия на 3 месяца (в/в и в ингаляциях), возможно, с пероральными антибиотиками. Можно Амоксициллин+клавулановая кислота (или Сульфаметоксазол и Триметоприм) в течение 1 месяца и ингаляции Колистиметата натрия в течение 3 месяцев.
2. При хронической инфекции длительно ингаляции Колистиметата натрия (1-я линия), при отсутствии эффекта ингаляции Меропенема (2-я линия).
3. Целесообразна комбинация двух антисинегнойных антибиотиков различных классов (табл. 17). (УД – С).
Таблица – 20. Антибиотики, применяемые при высеве Burkholderia cepaciacomplex, Achromobacter xylosoxidans [1–3, 6, 9, 12, 15-19, 33, 36, 38, 46, 51, 62-68]:
| Антибиотик | Доза в сутки для детей | Суточные дозы для взрослых | Путь введения |
Число приемов в день |
| Цефтазидим | 300 мг/кг | 9-12 г | в/в | 3 |
| Цефтазидим | В возрасте до 2 мес: 25-50 мг/кг/сут, старше 2 мес: 50-100 мг/кг/сут | 2 г | ингаляции | 2 |
| Меропенем | 120 мг/кг | 6г | В/в | 3 |
| Меропенем | 250 мг -500 мг | 250 мг -500 мг | Ингаляции | 2 |
| Пиперациллин+ Тазобактам | 400-500 мг/кг | 13,5 | В/в | 3 |
| Сульфаметоксазол и Триметоприм | 20мг/кг (по триметоприму) | 2880мг | В/в и внутрь | 3 |
| Доксициклин (старше 12 лет) | 100-200 мг | 1 день-200 мг затем 100 мг | Внутрь | 1 |
| Хлорамфеникол | 50-100 мг/кг | 2 – 4 г | Внутрь В/в | 3-4 |
АБТ при высеве из бронхиального секрета Stenotrophomonas maltophilia.
Распространенность Stenotrophomonas maltophilia варьирует, достигая в центрах МВ 25% [69]. Хроническая инфекция Stenotrophomonas maltophilia — предиктор частых обострений легочной инфекции [70].
Таблица 21. Антибиотики, применяемые при высеве Stenotrophomonas spp.
| Антибиотик | Доза в сутки для детей | Суточные дозы для взрослых | Путь введения |
Число приемов в день |
| Сульфаметоксазол и Триметоприм | 20 мг/кг (по триметоприму) | 2880 мг | В/в, внутрь | 2-3 |
| Хлорамфеникол | 50-100 мг/кг | 2-4 г | Внутрь | 3-4 |
| Цефтазидим | 150 мг/кг | 9 г | В/в | 3 |
| Цефтазидим | 2 г/сут | 2 г | Ингаляции | 2 |
| Пиперациллин/Тазобактам | 400 мг/кг | 13,5 г | В/в | 4 |
| Ципрофлоксацин | 30 мг/кг | 800 мг | В/в | 2 |
| Ципрофлоксацин | 50 мг/кг | 1,5-2 г | Внутрь | 2 |
1) Stenotrophomonas maltophilia характеризуется отсутствием чувствительности к карбапенемам, высокой резистентностью к азтреонаму, АМГ, тазобактаму, колистиметату натрия.
2) Наиболее активен Сульфаметоксазол и Триметоприм.
3) При легких проявлениях возможно пероральное назначение АБП (Сульфаметоксазол и Триметоприм, хлорамфеникол).
4) Рациональна комбинация Сульфаметоксазол и Триметоприма с цефтазидимом.
5) Курс терапии составляет 2-4 недели.
Противовоспалительная терапия.
Наибольшее влияние на продолжительность жизни больных МВ оказывают инфекционные осложнения со стороны органов дыхания. Характерной особенностью легочной болезни при МВ является бурная воспалительная реакция, сопровождающаяся повышенной продукцией провоспалительных цитокинов и выраженной нейтрофильной инфильтрацией. В связи с этим противовоспалительная терапия при МВ актуальна [78-86].
В качестве противовоспалительных препаратов применяются:
- макролидные антибиотики (азитромицин или кларитромицин);
- нестероидные противовоспалительные препараты (ибупрофен);
- системные и ингаляционные кортикостероиды.
Макролидные антибиотики используются с противовоспалительной целью при МВ в сочетании с хронической синегнойной инфекцией [80-82, 86, 87]. Макролиды снижают образование провоспалительных цитокинов, при длительном применении в субтерапевтических дозах обладают прямым противовоспалительным эффектом, уменьшают продукцию провоспалительных цитокинов TNF-α и IL-8; подавляют внутрилегочный выброс нейтрофилов и нейтрофильную хемотаксическую активность, затрудняют адгезию P.aeruginosa к слизистой бронхов [80-82]. В комбинации с фторхинолонами (ципрофлоксацин) усиливают действие последних за счет улучшения проникновения внутрь микробной клетки; оказывают прямое воздействие на P.aeruginosa в виде уменьшения ее жизнеспособности,. Благодаря системному противовоспалительному эффекту длительный прием макролидов существенно снижает частоту гепатобилиарных осложнений (УД В) [86, 87]. В качестве системных противовоспалительных препаратов применяют азитромицин и кларитромицин.
Основным показанием к применению макролидных антибиотиков является наличие хронической синегнойной инфекции.
Методика назначения:
- азитромицин назначается в дозе 250 мг (больным с весом менее 40 кг) и 500 мг (больным с весом 40 кг и более) через два дня на третий между приемами пищи; длительность терапии индивидуальна;
- кларитромицин в дозе 125 мг (больным с весом менее 40 кг) и 250 мг (больным с весом 40 кг и более) через день независимо от приема пищи [86].
Нестероидные противовоспалительные препараты (НПВП) при МВ рассматриваются как альтернатива ГКС (только ибупрофен). Высокие дозы ибупрофена (концентрация в плазме 50-100 г/мл) продемонстрировали сохранение функции легких, замедление прогрессирования поражения легких при МВ, [83], риск побочных эффектов связан в основном с желудочно-кишечными расстройствами и кровотечением, при этом польза превалирует над рисками (УД В) [83, 89].
Методика назначения: ибупрофен в дозе 20-30 мг на 1 кг массы тела 2 раза в день детям в возрасте от 6 лет и взрослым; максимальная суточная доза для взрослых составляет 1,2 г; для детей и подростков в возрасте от 12 до 17 лет – 1,0 г.
Глюкокортикостероиды (ГКС) обладают высокой системной противовоспалительной активностью. Использование ГКС в ряде случаев является необходимым, единственным решением при МВ. Пероральные ГКС улучшают функцию легких (при дозе преднизолона 1-2 мг/кг с длительностью применения до 4-5 лет), их использование при МВ ограничивается серьезными побочными эффектами [84]:
- частые, неопасные для здоровья и жизни, дозозависимые: экзогенный гиперкортицизм (увеличение аппетита, прибавка веса, кожно-трофические изменения, истончение, сухость кожи, стрии, угри, усиление капиллярного рисунка), лейкоцитоз, гипокалиемия, гепатомегалия;
- нечастые, зависят от индивидуальных особенностей, генетической и конституциональной предрасположенности: подавление функции коры надпочечников и продукции АКТГ, инфекционные осложнения, повышение АД, остеопороз, язвенный процесс в желудочно-кишечном тракте, гипергликемия и гликозурия, психические расстройства, миопатия, катаракта, задержка роста.
Для минимизации данных эффектов используются ингаляционные кортикостероиды (ИГКС) [84, 86, 87].
Показания к применению ИГКС: сочетание МВ с бронхиальной астмой, гиперреактивностью бронхов, наличие аллергического ринита. Применяют беклометазона дипропионат, будесонид, флутиказона пропионат в форме монотерапии или в комбинации с β2-агонистами различной длительности действия (формотерол, салметерол, сальбутамол). Используются дозы, рекомендованные при данных заболеваниях в различных видах ингаляционного применения.
Пероральное применение ГКС при легочной и смешанной формах МВ может быть рекомендовано при [84, 86, 87]:
- тяжелое течение МВ с частыми обострениями, выраженной дыхательной недостаточностью;
- обструктивный синдром, рефрактерный к действию β2-агонистов;
- при воспалении с образование ателектазов в легких;
- аллергический бронхолегочный аспергиллез (АБЛА).
Методика назначения преднизолона: из расчета 1 (1-2) мг/кг фактического веса, внутрь в течение 15-20 дней; скорость снижения определяется исходной дозой: при 15 мг/сут и более снижение по 1-1,25 мг 1 раз в 3-4 дня; с 15-10 мг/сут снижение по 1-1,25 мг 1 раз в 5-7 дней; при необходимости более длительной терапии — альтернирующий курс приема преднизолона.
Таблица 22. Перечень основных лекарственных средств (имеющих 100% вероятность применения):
| Фармакотерапевтическая группа | Международное непатентованное наименование ЛС | Способ применения | Уровень доказательности |
| Цефалоспорин | Цефепим 1г | в/в | С |
| Цефтазидим 1 г | в/в | С | |
| Цефтриаксон1 г | в/в | С | |
| Фторхинолон | Ципрофлоксацин 200 мг/100 мл, 250-500 мг | в/в, per os | С |
| Сульфаниламид | Сульфаметоксазол и Триметоприм120 мг, 480 мг | в/в, per os | С |
| Макролид | Азитромицин 50 мг, 125 мг, 250 мг, 500 мг; 100 мг/5 мл; |
per os в/в |
С |
| Карбапенем | Меропенем 500 мг / 1,0 г | в/в | С |
| Пенициллины комбинированные | Пиперациллин и тазабактам 2,5 г | в/в | С |
|
Аминогликозид |
Тобрамицин 300мг/5мл |
раствор для ингаляции | С |
|
Тобрамицин 112 мг (4 капсулы по 28 мг) |
порошок для ингаляций | С | |
| Антибактериальный препарат других групп | Колистиметат натрия 2000 000 МЕ | раствор для ингаляций | С |
| Муколитики | |||
| Ацетилцистеин | 400 мг/2 мл | раствор для ингаляций | С |
| Дорназа– альфа |
2,5 мг/2,5 мл |
для ингаляции | С |
| 3-5-7%NaCl | 100,0мл | для ингаляции | С |
| Ферменты | |||
| Панкреатин |
10 000ЕД; 25 000 ЕД; |
Рer os | С |
| Противогрибковые препараты | |||
| Флуконазол |
2мг/мл, 50 мг, 100 мг, 150 мг |
per os | С |
| Вориконазол |
200 мг, 50 мг |
рer os, раствор для инфузий | С |
| Каспофунгин | 50 мг | для инфузий | С |
| Амфотерицин В | 50мг, 10мл | для инфузий | С |
| Микафунгин | 50 мг | для инфузий | С |
| Позаконазол | 40 мг/мл | per os | С |
| Бронхолитики | |||
| Сальбутамол |
100 мкг/доза; 5 мг/мл; |
Аэрозоль, раствор для ингаляций | С |
| Ипротропия бромид | 0,025%, 250мкг/мл | Аэрозоль, раствор для ингаляций | С |
| Фенотерол/ипротропия бромид |
0,5/0,25 мг в 1 мл 0,5/0,2 мг в дозе |
Аэрозоль, раствор для ингаляций | С |
| Глюкокортикостероиды | |||
| Преднизолон |
30 мг/мл, 5 мг |
per os, в/м,в/в | С |
| Будесонид | 0,25-0,5 мг/мл | раствор для ингаляций | С |
Таблица 23. Перечень дополнительных лекарственных средств (имеющие менее 100% вероятности применения):
|
Фармакотерапевти ческая группа |
Международное непатентованное наименование ЛС | Способ применения | Уровень доказательности |
| Монобактам | Азтреонам 500 мг, 1000 мг | в/в | С |
| Карбапенем в комбинации |
Имипенем+ Циластатин |
в/в | С |
| Тетрациклины | Доксициклин 50 мг, 100 мг | внутрь | С |
| Амфеникол | Хлорамфеникол 250 мг, 500 мг; 0,5 г, 1,0 г |
внутрь в/в |
С |
| цефалоспорин+бета-лактамаз ингибитор |
Цефоперазон+ Сульбактам 1,0 г |
в/в | В |
| Витамины | |||
| Токоферол | 100 мг | для приема внутрь | С |
| Холекальциферол |
15000 МЕ/мл; |
для приема внутрь | С |
| Эргокальциферол | 0,125 % | для приема внутрь | С |
| Ретинол ацетат | 5000 МЕ, 33000 МЕ. | для приема внутрь | С |
Лечение (стационар)
ТАКТИКА ЛЕЧЕНИЯ НА СТАЦИОНАРНОМ УРОВНЕ
Немедикаментозное лечение: см. Амбулаторный уровень
Медикаментозное лечение: см. Амбулаторный уровень
Осложнения при муковисцидозе: остеопороз, сахарный диабет.
Остеопороз при МВ [1, 71] носит вторичный характер. Основным клиническим проявлением остеопороза при МВ, являются остеопоротические переломы костей при незначительной травме или спонтанно; любой локализации, часты переломы тел позвонков, проксимальных отделов бедренной и плечевой костей, дистального отдела предплечья.
Диагностика остеопороза проводится по клинической картине, а также дополнительным лабораторно-инструментальным данным [1, 24, 26-28,71]:
— Остеоденситометрия — «золотой стандарт». В возрасте до 20 лет зоны измерения — поясничный отдел позвоночника и все тело, исключая голову.
Первое рутинное исследование костной плотности у детей рекомендовано в возрасте 8-10 лет, но может быть раньше при наличии факторов риска остеопороза [71]. Повторные измерения МПК проводят: • каждые 5 лет (если Z-критерий выше -1 SD), каждые 2 года (Z-критерий между -1 и -2 SD) или ежегодно (Z-критерий < -2 SD).
— Рентгенография костей скелета. У детей и подростков основное показание для проведения рентгенографии грудного и поясничного отделов позвоночника — длительная терапия пероральными глюкокортикоидами [25-28]. У взрослых — боли в спине, длительный прием системных ГКС, грудной кифоз.
— Лабораторные методы исследования, в крови: общий и ионизированный кальций, фосфор, креатинина, щелочную фосфатазу, содержания витамина D (25(OH)D). маркеры костного обмена: P1NP (маркер костеообразования) и СТХ (маркер резорбции кости)
Профилактика остеопороза при МВ: сбалансированное питание, физическая активность, лечение основного и сопутствующих (например, сахарный диабет) заболеваний, нормализация ИМТ, адекватное содержание витамина D, кальция и других питательных веществ (белки, калий, магний, медь, железо, фтор, цинк, витамины А, С и К).
Лечение остеопороза при МВ проводится согласно существующим рекомендациям (в т.ч. с применением бифосфонатов и др. субстанций, рекомендованных при остеопорозе).
Муковисцидоз-ассоциированный сахарный диабет:
Сахарный диабет, обусловленный муковисцидозом (МЗСД) развивается постепенно, основные симптомы: полиурия, полидипсия, невозможность увеличения/поддержания массы тела, несмотря на увеличение питания, нарушение роста, задержка пубертата, ухудшение легочной функции [2].
Диагностика МЗСД: глюкоза, гликированный гемоглобин (НbА1с), оральный глюкозотолерантный тест (ОГТТ)
Лечение МЗСД: инсулинотерапия в зависимости от потребностей и клинической картины. Пероральные сахароснижающие препараты не рекомендованы.
Цирроз печени при МВ. Частота встречаемости заболеваний печени при МВ от 27 до 35%. У 5–10% пациентов с МВ в течение первого десятилетия жизни развивается цирроз печени [1, 72]. Большинству пациентов с циррозом печени при МВ требуется трансплантация печени при прогрессировании портальной гипертензии [72, 73]. Проведение ранней трансплантации печени приводит к стабилизации/улучшению функции легких после операции [74].
Хирургическое вмешательство [2]:
— при необходимости установки гастростомы;
— установка венозного порта для длительной повторной внутривенной антибактериальной терапии;
— при осложнениях со стороны дыхательной системы (пневмоторакс, легочное кровотечение и др.), со стороны желудочно-кишечного тракта;
— При декомпенсации респираторной функции (хроническая гиперкапния, высокая легочная гипертензия) — решение вопроса о трансплантации легких [75-77], Трансплантация легких показана при терминальном поражением легких, с тяжелой дыхательной недостаточностью, выраженных функциональных ограничениях (3-4-й ФК по NYHA), с риском смерти в ближайшие 2 года более 50%.
Показания к трансплантации легких при МВ:
• снижение ОФВ1<30% от д.в., несмотря на проводимую в полном объеме медикаментозную терапию, с наличием инфицирования дыхательных путей нетуберкулезными микобактериями (в особенности Mycobacterium abscessus) или Burkholderia cepacia complex и/или наличием сахарного диабета;
• дистанция в тесте 6-минутной ходьбы менее 400 метров;
• легочная гипертензия в отсутствие гипоксемической дыхательной недостаточности;
• учащения эпизодов обострения заболевания, ассоциированных с любым из следующих состояний:
– острая дыхательная недостаточность, требующая НИВЛ;
– повышение антибиотикорезистентности или неудовлетворительное восстановление общеклинического состояния после обострения заболевания;
– ухудшение нутритивного статуса, несмотря на дополнительное питание;
– пневмоторакс;
– жизнеугрожающее кровохарканье, несмотря на эмболизацию бронхиальных артерий;
• хроническая дыхательная недостаточность:
– изолированная гипоксемическая форма (РО2 менее 60 мм рт.ст.);
– гиперкапническая форма (РСО2 более 50 мм рт.ст.);
• длительная (амбулаторная) неинвазивная вентиляция легких;
• выраженное ограничение функционального класса (4-й класс по NYHA).
Трансплантация печени при развитии цирроза печени у пациентов с МВ.
Факторы, влияющие на оценку сроков трансплантации печени [74]:
- Степень варикозного расширения вен пищевода;
- Наличие в анамнезе эпизодов желудочно-кишечного кровотечения;
- Бактериологический диагноз;
- Прогрессирование ухудшения функции легких (ОФВ1/ФЖЕЛ <50%), рецидивирующие респираторные мультирезистентные бактериальные инфекции;
— Степень тромбоцитопении, лейкопении;
— Степень нарушения белково-синтетической функции печени, коагулопатия;
— Уровень цитолиза и холестаза;
— Артериальная легочная гипертензия;
— Наличие потенциального родственного донора печени;
— Прогрессирование ухудшения нутритивного статуса.
Показания для трансплантации печени при МВ:
- Цитолиз и/или холестаз в биохимических показателях крови;
- Снижение уровня лейкоцитов, тромбоцитов в крови ниже нормы;
- Изменения при УЗИ органов брюшной полости (изменение эхогенности и размеров печени, увеличение размеров селезенки, признаки портальной гипертензии);
- Наличие ВРВП по данным ЭГДС;
- Наличие коагулопатии, диспротеинемии.
Вакцинация пациентов с МВ.
Муковисцидоз — прямое показание для профилактических прививок в рамках Национального календаря без каких-либо отводов и отсрочек. МВ не является противопоказанием для прививок.
Дополнительно детей с МВ целесообразно прививать от менингококковой (с 2 лет), а также от гепатита А и ветряной оспы (с 12 мес), респираторной синцитиальновирусной инфекции, ротавирусной инфекции, ежегодно противогриппозная вакцинация.
Взрослых с МВ прививают по Национальному календарю от дифтерии, столбняка, кори, краснухи, гепатита В; по эпидемиологическим показаниям — против гепатита А, ветряной оспы, менингококковой инфекции, полиомиелита. Обязательна пневмококковая вакцинация с использованием пневмококковой полисахаридной конъюгированной тринадцативалентная вакцины однократно, а также ежегодно против гриппа.
Вакцинация при МВ проводится в т.ч. на фоне антибактериальной и иной терапии.
Дальнейшее ведение [31]:
Оказание медицинской помощи пациентам с МВ осуществляется на всех уровнях:
- Уровень первичной медико-санитарной помощи (ПМСП) по месту жительства пациента участковым врачом ВОП, педиатр, терапевт: контроль за общим состоянием, выписка рецептов, своевременное направление в городской/областной центры, республиканские центры муковисцидоза, при необходимости экстренная госпитализация по превалирующему поражению.
- городской/областной центры: консультации специалистов по необходимости (пульмонолог, гастроэнтеролог и др.), организация дневного стационара, стационара на дому, круглосуточного стационара, динамичная оценка лабораторно-инструментальных исследований, коррекция терапии, направление в республиканский центр муковисцидоза.
- республиканский центр муковисцидоза (как самостоятельная единица, на базе многопрофильного стационара с консультативно-диагностическим центром) оказывает специализированную, в т.ч. высокотехнологичную, медицинскую помощь (на амбулаторном и стационарном уровне), подбор и проведение реабилитационных программ и др.
Периодичность наблюдение пациентов с МВ:
- дети до 3-х мес – каждые 2 нед;
- 3-6 мес – 1 раз в мес;
- 6-12 мес – 1 раз в 2 мес;
- после 12 мес – ежеквартально, при необходимости чаще.
Таблица 24. Карта динамического наблюдения пациентов с МВ [31].
| ПМСП, областной уровень | |||
| № | Наименование исследования | Кратность исследования | Примечание |
| 1 | Антропометрия (рост, масса тела, расчет массо-ростового соотношения МРС) | 1 раз в 3 месяца | Динамическое наблюдение |
| 2 | Клинический анализ крови | 1 раз в 3 месяца | Динамическое наблюдение |
| 3 | Бактериологическое исследование мокроты (при невозможности собрать мокроту — мазок с задней стенки глотки) на микрофлору и чувствительность к антибиотикам | 1 раз в 3 месяца | Динамическое наблюдение |
| 4 | Функция внешнего дыхания (спирометрия) с 5 летнего возраста | 1 раз в 3 месяца | Динамическое наблюдение |
| 5 | Определение сатурации кислорода (пульсоксиметрия) | 1 раз в 3 месяца | Динамическое наблюдение |
| 6 | Рентгенограмма органов грудной клетки | 1 раз в год | Динамическое наблюдение |
| 7 | Биохимическое исследование крови (активность печеночных ферментов, соотношение белковых фракций, электролитный состав, 25(OH)D3, глюкоза и др.) | 1 раз в 6 месяцев | Динамическое наблюдение |
| 8 | Компьютерная томография органов грудной клетки | 1 раз в год | Динамическое наблюдение |
| 9 | Ультразвуковое исследование органов брюшной полости | 1 раз в год | Динамическое наблюдение |
| 10 | ЭКГ | 1 раз в год | Динамическое наблюдение |
| 11 | Эхо-КГ | 1 раз в год | Динамическое наблюдение |
| 12 | Копрологическое исследование | 1 раз в 3 месяца | Динамическое наблюдение |
| 13 | Фиброэзофагогастродуоденоскопия | (по показаниям) | Динамическое наблюдение |
| 14 | Компьютерная томография пазух носа | 1 раз в год | Динамическое наблюдение |
| 15 | Фибросканирование | (по показаниям) | Динамическое наблюдение |
| 16 | Консультация ЛОР — врача | 1 раз в год | Динамическое наблюдение |
| 17 | Консультация пульмонолога | 2 раза в год | Динамическое наблюдение |
| 18 | Консультация гастроэнтеролога | 1 раз в год | Динамическое наблюдение |
| 19 | Общий анализ мочи | 1 раз в 3 месяца | Динамическое наблюдение |
| 20 | Консультация эндокринолога | (по показаниям) | Динамическое наблюдение |
| 21 | Консультация кардиолога | 1 раз в год | Динамическое наблюдение |
| 22 | Консультация торакального хирурга | (по показаниям) | Динамическое наблюдение |
| 23 | Консультация абдоминального хирурга | (по показаниям) | Динамическое наблюдение |
| 24 | Контроль навыков кинезитерапии, использования дыхательных тренажеров и приборов | 1 раз в 3 месяца | Динамическое наблюдение |
| Республиканский уровень | |||
| Консультация координатора по муковисцидозу |
1 раз в год Повторно — при изменении тактики терапии |
Динамическое наблюдение Коррекция терапии |
|
| Определение хлоридов пота | 1 раз в год | Верификация диагноза | |
| Тест на толерантность к глюкозе | 1 раз в год |
Верификация диагноза Динамическое наблюдение |
|
| Активность эластазы в кале | 1 раз в год** |
Верификация диагноза Динамическое наблюдении |
|
| Молекулярно-генетический анализ – определение на ген муковисцидоза | 1 раз в год | Верификация диагноза |
Индикаторы эффективности лечения:
- Улучшение/стабильность общего состояние больного;
- Уменьшение (стабильная частота) частоты обострений;
- Улучшение/стабильность лабораторных показателей;
- Нормализация/стабильность показателей веса и роста;
- Отсутствие осложнений/отсутствие прогрессирования осложнений;
- Сокращение частоты экстренных госпитализаций;
- Сохранение качества жизни пациентов.
Госпитализация
ПОКАЗАНИЯ ДЛЯ ГОСПИТАЛИЗАЦИИ С УКАЗАНИЕМ ТИПА ГОСПИТАЛИЗАЦИИ [1-21]
Показания для плановой госпитализации с круглосуточным пребыванием:
- часто повторяющиеся синдром нарушенного кишечного всасывания неясного генеза в течение месяца и более;
- необходимость проведения дообследования в случае невозможности в амбулаторных условиях;
- подбор и коррекция терапии;
- необходимость проведения плановой или, при развитии нетяжелого обострения, внутривенной антибактериальной терапии при отсутствии возможности проведения ее в условиях дневного стационара или стационара на дому;
- необходимость планового оперативного вмешательства;
- установка венозных портов, гастростомы;
- необходимость оперативного лечения осложнений муковисцидоза (полипотомия, радикальная гайморотомия, спленэктомия, склерозирование вен пищевода и т.д.);
- трансплантация легких, печени.
Показания для экстренной госпитализации:
- впервые выявленный МВ;
- обострение муковисцидоза;
- развитие острой и/или быстрое прогрессирование хронической дыхательной недостаточности;
- легочное кровотечение, кровохарканье некупирующееся;
- пневмоторакс;
- кровотечение из варикозно-расширенных вен (ВРВ) пищевода, ВРВ верхних отделов желудка;
- признаки кишечной непроходимости;
- синдром потери солей (псевдо-Барттера синдром: гипокалиемия, гипонатриемия, гипохлоремия, алкалоз) тяжелой степени, требующий круглосуточного мониторинга электролитов, внутривенного введения электролитов;
- острый панкреатит и обострение хронического;
- учащение жирного стула, потеря в весе более 5%.
Показания для госпитализации по принципу стационарозамещающей помощи «дневной стационар»:
- необходимость проведения внутривенной антибактериальной терапии;
- легочная реабилитация;
- установка венозных портов, гастростомы.
Информация
Источники и литература
-
Протоколы заседаний Объединенной комиссии по качеству медицинских услуг МЗ РК, 2018
- 1) C.Castellani, AlistairJ.A.Duff, ScottC.Bell, HarryG.M.Heijerman, AnneManck, FelixRatjen, IsabelleSermeGaudelus, KevinW. Southern, JurgBarben, PatrikA.Flume, PavlaHodkova, N.Kashirskaya, M.N.Kirszenbaum, SueMadge, HelenOxley, BarryPlant, SarahJane Schwarzenberg, AlanR.Smyth, GiovanniTaccetti, ThomasO.F.Wagner, SusanR.Wolfe, PavelDrevinck. ECFS best practice guidelines: the 2018 revision. Journal of Cystic Fibrosis 17(2018) 153-178.
2) Национальный консенсус «Муковисцидоз: определение, диагностические критерии, терапия» Е.И. Кондратьева, Н.Ю. Каширская, Н.И. Капранов. Российское общество медицинских генетиков Российское респираторное общество Союз педиатров России Общероссийская общественная организация «Всероссийская ассоциация для больных муковисцидозом». Москва,2016г.
3) Красовский СА, Каширская НЮ, Черняк АВ и др. Генетическая характеристика больных муковисцидозом в Российской Федерации по данным Национального регистра (2014 г.). Пульмонология. 2016; 26 (2): 133-151.
4) Капранов Н.И., Каширская Н.Ю. Муковисцидоз. В кн.: Хронические заболевания легких у детей. Розинова Н.Н., Мизерницкий Ю.Л., ред. – М.: Практика, 2011: 94-107.
5) Munck A, Mayell SJ, Winters V, Shawcross A, Derichs N, Parad R, Barben J, Southern K W. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. JCystFibros. 2015; 14 (6): 706-713.
6) Федеральные клинические рекомендации по оказанию медицинской помощи детям с кистозным фиброзом (муковисцидозом). 2015г.
7) Ooi CY, Castellani C, Keenan K, Avolio J, Volpi S, Boland M, Kovesi T, Bjornson C, Chilvers MA, Mor-gan L, van Wylick R, Kent S, Price A, Solomon M, Tam K, Taylor L, Malitt KA, Ratjen F, Durie PR, Gons-ka T. Inconclusive diagnosis of cystic fibrosis after newborn screening. Pediatrics. 2015; 135 (6): 1377-1385.
 LaRusch J, Jung J, General IJ, Lewis MD, Park H W, Brand R E, Gelrud, A, Anderson M A, Banks PA, Conwell D, Lawrence C, Romagnuolo J, Baillie J, Alkaade S, Cote G, Gardner TB, Amann ST, Slivka A, Sandhu B, Aloe A, Kienholz ML, Yadav D, Barmada MM, Bahar I, Lee MG, Whitcomb DC. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLOS Genetics. 2014; 10 (7): e1004376 (1-15).
LaRusch J, Jung J, General IJ, Lewis MD, Park H W, Brand R E, Gelrud, A, Anderson M A, Banks PA, Conwell D, Lawrence C, Romagnuolo J, Baillie J, Alkaade S, Cote G, Gardner TB, Amann ST, Slivka A, Sandhu B, Aloe A, Kienholz ML, Yadav D, Barmada MM, Bahar I, Lee MG, Whitcomb DC. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLOS Genetics. 2014; 10 (7): e1004376 (1-15).
9) Муковисцидоз. Под редакцией Н.И. Капранова, Н.Ю. Каширской. МЕДПРАКТИКА-М.: 2014. 672 с.
10) Guidelines for the performance of the sweat test for the investigation of the CF in the UK. 2014. http://www.rcpch.ac.uk/child-health/standards-care/clinical-guidelines-and-standards/endorsed-and-supported/respiratory-med#ACB
11) Munck A, Mayell SJ, Winters V. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J Cyst Fibros. 2015; 14: 706–713.
12) Clinical Guidelines: Care of Children with Cystic fibrosis. www.rbht.nhs.uk/childrencf 2014 6th edition.
13) Standards of Care and Good Clinical Practice for the Physiotherapy Management of Cystic Fibrosis. Second edition. June 2011.
14) Consensus document outlining standards of care and food practice for physiotherapy. Laboratory Standards for Processing Microbiological Samples from People with Cystic Fibrosis. First edition. September 2010.
15) Standards for the Clinical Care of Children and Adults with Cystic Fibrosis in the UK. Second edition. December 2011.
16) Аntibiotic Treatment for Cystic Fibrosis (Report of the UK Cystic Fibrosis Trust Antibiotic Working Group, Thild Edition), Systic Fibrosis Trust, may 2009. – P.1,0 – 9,4.
17) Smyth АR, Bell SC, Bojcin S, Bryon M, Duff A, Flume P, Kashirskaya N, Munck A, Ratjen F, Schwarzenberg SJ, Sermet-Gaudelus I, Southern KW, Taccetti G, Ullrich G, Wolfe S. European cystic fibrosis society standarts of care working group. Best practice guidelines. J Cyst Fibros. 2014; 13 (1): 23-42. https://www.ecfs.eu/ecfs-standards-care/references.
18) P.J. Mogayzel, E.T. Naureckas, K.A. Robinson Cystic Fibrosis Pulmonary Guidelines. Am. J. Respir. Crit. Care Med. 2013; 187: 680–689.
19) M. Proesmans, F. Vermeulen, L. Boulanger, J. Verhaegen, K. De Boeck. Comparison of two treatment regimens for eradication of Pseudomonas aeruginosa infection in children with cystic fibrosis. J Cyst Fibros. 2013; 12(1):29-34.
20) Регистр больных муковисцидозом в Российской Федерации. 2014 год. – М.: Медпрактика-М, 2015.
21) LittlewoodJM, WolfeSP. Control of malabsorption in cystic fibrosis. Paediatr Drugs. 2000; 2 (3): 205-22. 13. Turck D, Braegger CP, Colombo C, Dimitri Declercq D, Morton A, PanchevaR, Robberecht E, Stern M, Wolfe S, Schneider SM, Wilchansky M. ESPEN-ESPGHAN guidelines on nutrition care for infants, children and adults with cystic fibrosis. Clinical Nutrition 2016; 35: 557-577.
22) Красовский С.А. Остеопороз у взрослых больных муковисцидозом. Автореф. дис. … канд. мед. наук. М., 2012.
23) Соболенкова В.С. Системный анализ в ранней диагностике и лечении остеопенического синдрома при муковисцидозе. Автореф. дис. … канд. мед. наук. Тула, 2009.
24) ArisRM, MerkelPA, BachrachLK, BorowitzDS, BoyleMP, ElkinSL, GuiseTA, Hardin DS, Haworth CS, HolickMF, JosephPM, O’BrienK, TullisE, WattsNB, WhiteTB. Consensus statement: Guide to bone health and disease in cystic fibrosis. J. Clin. Endocrinol. Metab. 2005; 90(3): 1888-96.
25) Ашерова И.К. Снижение тяжести течения заболевания, повышение выживаемости и качества жизни больных муковисцидозом на основе совершенствования междисциплинарной специализированной помощи. Автореф. дис. … докт. мед. наук. М., 2013.
26) Капранов Н.И., Капустина Т.Ю. Состояние минеральной плотности костной ткани у пациентов с муковисцидозом. Педиатрия. 2008; 5: 36-41.
27) Симанова Т.В. Клинико-генетические особенности и костный метаболизм у больных муковисцидозом. Автореф. дис. … канд. мед. наук. М., 2009.
28) Горинова Ю.В. Остеопения при хронических болезнях легких у детей. Автореф. дис. … канд. мед. наук. М., 2005.
29) Консенсус ISPAD по клинической практике (РагнарХанас, Ким С. Донахью, Джорджианна Клин- генсмит, Питер Д.Ф. Свифт), 2009. С. 233.
30) Клинические рекомендации «Алгоритмы медицинской помощи больным сахарным диабетом». 7-й вып. Под ред. И.И. Дедова, М.В. Шестаковой. 2015. С. 112.
31) Методические рекомендации «Алгоритм ранней диагностики некоторых редких заболеваний у детей» (мукополисахаридозы, болезнь Гоше, муковисцидоз и первичные иммунодефициты). /Р.З.Боранбаева, М.Н.Шарипова, Г.К.Абдилова, Л.Н.Манжуова, Ш.Т.Наурызалиева // Алматы: Научный центр педиатрии и детской хирургии,2017.-28с.
32) Клинические рекомендации: Медицинский уход за детьми с муковисцидозом. Королевская больница Бромптонэнд Харфилд, 6-еиздание, 2014.
33) Clinical Guidelines: Care of Children with Cystic Fibrosis Royal Brompton Hospital,7th edition, 2017.
34) World Health Organization. Classification of cystic fibrosis and related disorders, Report of a Joint Working Group of WHO/ICF(M)A/ECFS/ECFTN, 2001 (reprinted in J Cyst Fibros. 2002; 1: 5-8).
35) Bombieri C., M.Claustres, K.De Boeck, N.Derichs, J.Dodge at al. Recommendations for the classification of diseases as CFTR-related disorders / 2011 European Cystic Fibrosis Society // Journal of Cystic Fibrosis Volume 10 Suppl 2 (2011) S86–S102
36) Lambiase A, Catania MR, Del Pezzo M, Rossano F, Terlizzi V, Sepe A, Raia V. Achromobacter spp. respiratory tract infection in cystic fibrosis patients. Eur. J. Clin. Microbiol. Infect. Dis. 2011 Aug; 30 (8): 973-980.
37) Rogers GB, Carroll MP, Serisier DJ, Hockey PM, Jones GR, Bruce KD. Characterization of bacterial community diversity in cystic fibrosis lung infections by use of 16S ribosomal DNA terminal restriction fragment length polymorphism profiling. J Clin Microbiol 2004; 42: 5176–5183.
38) Tunney MM, Field TR, Moriarty TF, Patrick S, Doering G, Muhlebach MS, Wolfgang MC, Boucher R, Gilpin DF, McDowell A, Elborn JS. Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med 2008; 1;177(9):995- 1001.
39) C. Williams, R. Ranjendran, G. Ramage Pathogenesis of Fungal Infections in Cystic Fibrosis. Curr Fungal Infect Rep 2016; 10:163–169.
40) M. Pihet, J. Carrere, B. Cimon, D. Chabasse, L. Delhaes, F. Symoens, J. P. Bouchara. Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis—a review. Medical Mycology 2009;
41) S.Ziesing, S.Suerbaum, L.Sedlace. Fungal epidemiology and diversity in cystic fibrosis patients over a 5-year period in a national reference centerMedical Mycology 2016; 54: 781–786
42) M.Sinaasappel, M.Stern, J.Littlewood, S.Wolfe, G.Steinkamp, HarryG.M. Heijerman. Nutrition in patients with cystic fibrosis: a European Consensus. J Cyst Fibrosis. 2002 (1): 51–75.
43) Borowitz D, Robinson KA, Rosenfeld M, Davis SD, Sabadosa KA, Spear SL, Michel SH, Parad RB, White TB, Farrel PM, Marshall BC, MD, Accurso FJ. Cystic fibrosis foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. 2009; 155: 73-93.
44) Рославцева Е.А., Капранов Н.И., Каширская Н.Ю., Симонова О.И. Национальная программа оптимизации вскармливания детей первого года жизни. M.: Союз педиатров России, 2011: 48, 49.
45) Turck D, Braegger CP, Colombo C, Dimitri Declercq D, Morton A, Pancheva R, Robberecht E, Stern M, Wolfe S, Schneider SM, Wilchansky M. ESPEN-ESPGHAN guidelines on nutrition care for infants, children and adults with cystic fibrosis. Clinical Nutrition. 2016; 35: 557-77.
46) P.A. Flume, P.J. Mogayzel, K.A. Robinson Cystic Fibrosis Pulmonary Guidelines: Treatment of Pulmonary Exacerbations. Am. J. Respir. Crit. Care Med. 2009; 180: 802–808.
47) Donaldson S.H., Bennett W.D., Zeman K.L., Knowles M.R., Tarran R., Boucher R.C. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N. Engl. J. Med. 2006; 354: 241–50.
48) Wark P.A., McDonald V., Jones A.P. Nebulised hypertonic saline for cystic fibrosis. Cochr. Database Syst. Rev. 2005 (3): CD001506. Doi: 10.1002/14651858.
49) Ros M., Casciaro R., Lucca F., Troiani P., Salonini E., Favilli F., Quattrucci S., Sher D., Assael B.M. Hyaluronic Acid improves the tolerability of hypertonic saline in the chronic treatment of cystic fibrosis patients: a multicenter, randomized, controlled clinical trial. J Aerosol Med Pulm Drug Deliv. 2014; 27 (2): 133–7.
50) Bilton D., Robinson P., Cooper P., Gallagher C.G., Kolbe J., Fox H., Jaques A, Chariton B. Inhaled dry powder mannitol in cystic fibrosis: an efficacy and safety study. Eur. Respir. J. 2011; 38: 1071–80.
51) Орлов А.В., Симонова О.И., Рославцева Е.А., Шадрин Д.И. Муковисцидоз (клиническая картина, диагностика, лечение, реабилитация, диспансеризация): учебное пособие для врачей. СПб.: Изд-во СЗГМУ им. И. И. Мечникова, 2014. — 160 с.
52) Клинический протокол медицинского вмешательства «Виброакустичекая терапия». Утвержден Экспертным советом МЗ РК Протокол №__ от _________ г.
53) McCool FD, Rosen MJ. Nonpharmacologic Airway Clearance Therapies; ACCP Evidence-Based. Chest. 2006:129:250-259.
54) Osman LP, Roughton M, Hodson ME, Pryor JA. High frequency chest wall oscillation in cystic fibrosis. Journal of Cystic Fibrosis 7; Supplement 2: S73, 295, 2008.
55) Pryor JA, Prasad SA. Physiotherapy Techniques in: Pryor JA, Prasad SA (Eds). Physiotherapy for respiratory and cardiac problems (4th edn). Churchill Livingstone, Edinburgh pp 134 — 217, 2008.
56) Darbee JC, Ohtake PJ, Grant BJ, Cerny FJ. Physiological evidence for the efficay of positive expiratory pressure as an airway clearance technique in patients with cystic fibrosis. Phys Ther 2004;84(6):524-37.
57) Brooks D, Newbold E, Kozar LF, Rivera M. The flutter device and expiratory pressures. J Cardiopulm Rehabil. 2002 Jan-Feb; 22(1):53-7.
58) Finck BJ. Forced expiration technique, directed cough and autogenic drainage. Respir Care 2007;52;9: 1210- 1223.
59) Calvo Lerma J, Hulst J, Asseiceira I, Claes I, Garriga M, Colombo C, Walet S., Martins T, Boon M, Ruperto M, Speziali C, Woodcock S, Witters P, Masip E, Barreto C, de Boeck C, Ribes-Koninckx C. Nutritional status, nutrients intake and enzymatic supplements in a European Cystic Fibrosis cohort: a cross-sectional overview. J Cyst Fibros. 2016; 15 (1): 3.
60) Gooding, D.Westaby. Gastrointestinal disease in cystic fibrosis. In Cystic fibrosis. Third ed. Hodson M, Duncan G, Bush A (eds.). London: Edward Arnold (Publishers) Ltd., 2007.
61) Heubi JE, Schaeffer D, Ahrens RC, Sollo N, Strausbaugh S, Graff G, Jain R, Witte S, Forssmann K. Safety and efficacy of a novel microbial lipase in patients with exocrine pancreatic insufficiency due to cystic fibrosis: a randomized controlled clinical trial. J Pediatr. 2016; 176: 156-61.
62) F. Ratjen, A. Munck, P. Kho, G. Angyalosi Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis: the ELITE trial. Thorax. 2010 ;65 (4): 286- 291.
63) G. Taccetti, E. Bianchini, L. Cariani. Early antibiotic treatment for Pseudomonas aeruginosa eradication in patients with cystic fibrosis: A randomised multicentre study comparing two different protocols. Thorax. 2012; 67 (10) :853-859. 48
64) Guss AM, Roeselers G, Newton ILG, et al. Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. The ISME Journal. 2011; 5 (1): 20–9.
65) Mayer-Hamblett N, Kronmal RA, Gibson RL, et al. Initial Pseudomonas aeruginosa treatment failure is associated with exacerdations in cystic fibrosis. Pediatr. Pulmonol. 2012; 47 (2): 125–34.
66) Mann HJ, Canafax DM, Cipolle RJ, et al. Increased dosage requirements of tobramycin and gentamicin for treating Pseudomonas pneumonia in patients with cystic fibrosis. Pediatr. Pulmonol. 1985; 1: 238–43.
67) De Groot R, Hack BD, Weber A, et al. Pharmacokinetics of ticarcillin in patients with cystic fibrosis: a controlled prospective study. Clin.Pharmacol.Ther. 1990; 47:73–8.
68) De Groot R, Smith AL. Antibiotic pharmacokinetics in cystic fibrosis. Differences and clinical significance. Clin. Pharmacokinet. 1987; 13: 228–53.
69) De Vrankrijker AMM, Wolfs TFW, Van der Ent CK. Challenging and emerging pathogens in cystic fibrosis. Paediatr. Respir. Rev. 2010; 11 (4): 246–54.
70) Waters V, Yau Y, Prasad S, et al. Stenotrophomonas maltophilia in cystic fibrosis: serologic response and effect on lung disease. Am. J. Respir. Crit. Care Med. 2011; 183: 635–40.
71) Sermet-Gaudelus I, Bianchi ML, Garabédian M, Aris RM, Morton A, Hardin DS, Elkin SL, Compston JE, Conway SP, Castanet M, Wolfe S, Haworth CS. European cystic fibrosis bone mineralisation guidelines. J Cyst Fibros. 2011; 10 Suppl 2: S16-23.
72) Debray D. et al. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease // J. Cyst. Fibros. 2011. Vol. 10. P. S29–S36.
73) Elborn J.S. Cystic fibrosis // Lancet. 2016. Vol. 388, № 10059. P. 2519–2531.
74) Milkiewicz P. et al. Transplantation for cystic fibrosis: outcome following early liver transplantation // J. Gastroenterol. Hepatol. 2002. Vol. 17, № 2. P. 208–213.
75) Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb SB, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Lung and Heart-Lung Transplantation Report-2015; Focus Theme: Early Graft Failure. J Heart Lung Transplant 2015 Oct 10;34(10):1264-77.
76) Benden C, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: sixteenth official pediatric lung and heart-lung transplantation report – 2013; focus theme: age. J Heart Lung Transplant 2013; 32: 989-997.
77) Lars H. Lund, Leah B. Edwards, Anne I. Dipchand et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-third Adult Heart Transplantation Report. 2016; Focus Theme: Primary Diagnostic Indications for Transplant. The Journal of Heart and Lung Transplantation. Vol 35, No 10, October 2016: 1170-1184.
78) Massimo Conese, Mario Romano, Maria Lucia Furnari, Elena Copreni, Ida De Fino, Francesca Pardo, Luis V. J. Galietta. New Genetic and Pharmacological Treatments for Cystic Fibrosis. Current Pediatric Reviews, 2009, 5: 8-27.
79) Balfour-Lynn IM, Lees B, Hall P, et al. Multicenter randomized controlled trial of withdrawal of inhaled corticosteroids in cystic fibrosis. Am J Respir Crit Care Med. 2006; 173: 1356-62.
80) Ianaro A, Ialenti A, Maffia P, et al. Anti-inflammatory activity of macrolide antibiotics. J Pharmacol Exp Ther 2000; 292: 156-63.
81) Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003; 290: 1749-56.
82) Pukhalsky AL, Shmarina GV, Kapranov NI, Kokarovtseva SN, Pukhalskaya D, Kashirskaja NJ. Antiinflammatory and immunomodulating effects of clarithromycin in patients with cystic fibrosis lung disease. Mediators Inflamm 2004; 13: 111-7.
83) Lands LC, Milner R, Cantin AM, Manson D, Corey M. High-dose ibuprofen in cystic fibrosis: Canadian safety and effectiveness trial. J Pediatr. 2007; 151: 249-54.
84) Ross KR, Chmiel JF, Konstan MW. Paediatr Drugs. The role of inhaled corticosteroids in the management of cystic fibrosis. 2009; 11 (2): 101-13.
85) Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Guideline Summary/ NGC:009821, 2013 Apr 1 https://www.guideline.gov/ summaries/summary/45307.
86) Cystic brosis pulmonary guidelines: chronic medications for main-tenance of lung health. Am J Respir Crit Care Med. 2007; 176: 957–69.
87) Cystic Fibrosis Pulmonary Guidelines, 2007; Oral steroids for long-term use in cystic fibrosis (Cochrane review), 2013: http://www.cochrane.org/CD000407/CF_use-oral-steroids-cystic-fibrosis)
88) Cheng K, Ashby D, Smyth RL Use of oral steroids for cystic fibrosis Published Online: June 24, 2013, http://summaries.cochrane.org/CD000407/use-of-oral-steroids-for-cystic-fibrosis
89) Schluchter MD, Konstan MW, Xue L, Davis PB. Relationship between high-dose ibuprofen use and rate of decline in FEV1 among young patients with mild lung disease in the CFF Registry. Ped. Pulmunol.2004; Suppl. 27: 322.
90) Kalnins D, Wilschanski M. Maintenance of nutritional status in patients with cystic fibrosis: new and emerging therapies. Drug Design, Development and Therapy 2012; 6: 151-61.
- 1) C.Castellani, AlistairJ.A.Duff, ScottC.Bell, HarryG.M.Heijerman, AnneManck, FelixRatjen, IsabelleSermeGaudelus, KevinW. Southern, JurgBarben, PatrikA.Flume, PavlaHodkova, N.Kashirskaya, M.N.Kirszenbaum, SueMadge, HelenOxley, BarryPlant, SarahJane Schwarzenberg, AlanR.Smyth, GiovanniTaccetti, ThomasO.F.Wagner, SusanR.Wolfe, PavelDrevinck. ECFS best practice guidelines: the 2018 revision. Journal of Cystic Fibrosis 17(2018) 153-178.
 LaRusch J, Jung J, General IJ, Lewis MD, Park H W, Brand R E, Gelrud, A, Anderson M A, Banks PA, Conwell D, Lawrence C, Romagnuolo J, Baillie J, Alkaade S, Cote G, Gardner TB, Amann ST, Slivka A, Sandhu B, Aloe A, Kienholz ML, Yadav D, Barmada MM, Bahar I, Lee MG, Whitcomb DC. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLOS Genetics. 2014; 10 (7): e1004376 (1-15).
LaRusch J, Jung J, General IJ, Lewis MD, Park H W, Brand R E, Gelrud, A, Anderson M A, Banks PA, Conwell D, Lawrence C, Romagnuolo J, Baillie J, Alkaade S, Cote G, Gardner TB, Amann ST, Slivka A, Sandhu B, Aloe A, Kienholz ML, Yadav D, Barmada MM, Bahar I, Lee MG, Whitcomb DC. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLOS Genetics. 2014; 10 (7): e1004376 (1-15).Информация
ОРГАНИЗАЦИОННЫЕ АСПЕКТЫ ПРОТОКОЛА
Список разработчиков протокола с указанием квалификационных данных:
- Боранбаева Риза Зулкарнаевна – доктор медицинских наук, директор АО «Научный центр педиатрии и детской хирургии».
- Наурызалиева Шамшагуль Тулеповна – кандидат медицинских наук, заведующая отделением пульмонологии АО «Научный центр педиатрии и детской хирургии».
- Садибекова Лейла Данигалиевна – кандидат медицинских наук, консультант отдела педиатрии КФ «UMC» «Национальный научный центр материнства и детства».
- Святова Гульнара Салаватовна – доктор медицинских наук, профессор, заведующая Республиканской медико-генетической консультацией АО «Научный центр акушерства, гинекологии и перинатологии».
- Мукатова Ирина Юрьевна – доктор медицинских наук, профессор кафедры кардиологии, внутренних болезней МСЭ и реабилитации НАО «Медицинский Университет Астана».
- Калиева Мира Маратовна – кандидат медицинских наук, доцент кафедры клинической фармакологии АО «Национальный Медицинский Университет».
Указание на отсутствие конфликта интересов: нет.
Рецензенты:
- Курманбекова Сауле Каспаковна – профессор кафедры педиатрии №2 АО «Национальный Медицинский Университет».
Условия пересмотра протокола: пересмотр протокола через 5 лет после его опубликования или при наличии новых сведений с уровнем доказательности.
Прикреплённые файлы
Мобильное приложение «MedElement»

- Профессиональные медицинские справочники. Стандарты лечения
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Мобильное приложение «MedElement»

- Профессиональные медицинские справочники
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Внимание!
Если вы не являетесь медицинским специалистом:
-
Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
-
Информация, размещенная на сайте MedElement и в мобильных приложениях «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта», не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
-
Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
-
Сайт MedElement и мобильные приложения «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта» являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
-
Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.