Not to be confused with the silicon-containing synthetic polymer silicone.
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Silicon | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Allotropes | see Allotropes of silicon | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Appearance | crystalline, reflective with bluish-tinged faces | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight Ar°(Si) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Silicon in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | group 14 (carbon group) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block | p-block | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Ne] 3s2 3p2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | 1687 K (1414 °C, 2577 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boiling point | 3538 K (3265 °C, 5909 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density (near r.t.) | 2.3290 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| when liquid (at m.p.) | 2.57 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | 50.21 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of vaporization | 383 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | 19.789 J/(mol·K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapor pressure
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | −4, −3, −2, −1, 0,[2] +1,[3] +2, +3, +4 (an amphoteric oxide) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.90 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionization energies |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic radius | empirical: 111 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | 111 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Van der Waals radius | 210 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Spectral lines of silicon |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | primordial | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | face-centered diamond-cubic
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Speed of sound thin rod | 8433 m/s (at 20 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal expansion | 2.6 µm/(m⋅K) (at 25 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | 149 W/(m⋅K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrical resistivity | 2.3×103 Ω⋅m (at 20 °C)[4] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Band gap | 1.12 eV (at 300 K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | diamagnetic[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar magnetic susceptibility | −3.9×10−6 cm3/mol (298 K)[6] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Young’s modulus | 130–188 GPa[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Shear modulus | 51–80 GPa[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bulk modulus | 97.6 GPa[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Poisson ratio | 0.064–0.28[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mohs hardness | 6.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 7440-21-3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Naming | after Latin silex or silicis, meaning ‘flint’ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Prediction | Antoine Lavoisier (1787) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery and first isolation | Jöns Jacob Berzelius[8][9] (1823) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Named by | Thomas Thomson (1817) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Isotopes of silicon
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| references |

Silicon is a chemical element with the symbol Si and atomic number 14. It is a hard, brittle crystalline solid with a blue-grey metallic luster, and is a tetravalent metalloid and semiconductor. It is a member of group 14 in the periodic table: carbon is above it; and germanium, tin, lead, and flerovium are below it. It is relatively unreactive.
Because of its high chemical affinity for oxygen, it was not until 1823 that Jöns Jakob Berzelius was first able to prepare it and characterize it in pure form. Its oxides form a family of anions known as silicates. Its melting and boiling points of 1414 °C and 3265 °C, respectively, are the second highest among all the metalloids and nonmetals, being surpassed only by boron.
Silicon is the eighth most common element in the universe by mass, but very rarely occurs as the pure element in the Earth’s crust. It is widely distributed in space in cosmic dusts, planetoids, and planets as various forms of silicon dioxide (silica) or silicates. More than 90% of the Earth’s crust is composed of silicate minerals, making silicon the second most abundant element in the Earth’s crust (about 28% by mass), after oxygen.
Most silicon is used commercially without being separated, often with very little processing of the natural minerals. Such use includes industrial construction with clays, silica sand, and stone. Silicates are used in Portland cement for mortar and stucco, and mixed with silica sand and gravel to make concrete for walkways, foundations, and roads. They are also used in whiteware ceramics such as porcelain, and in traditional silicate-based soda-lime glass and many other specialty glasses. Silicon compounds such as silicon carbide are used as abrasives and components of high-strength ceramics. Silicon is the basis of the widely used synthetic polymers called silicones.
The late 20th century to early 21st century has been described as the Silicon Age (also known as the Digital Age or Information Age) because of the large impact that elemental silicon has on the modern world economy. The small portion of very highly purified elemental silicon used in semiconductor electronics (<10%[citation needed]) is essential to the transistors and integrated circuit chips used in most modern technology such as smartphones and other computers. In 2019, 32.4% of the semiconductor market segment was for networks and communications devices, and the semiconductors industry is projected to reach $726.73 billion by 2027. [10]
Silicon is an essential element in biology. Only traces are required by most animals, but some sea sponges and microorganisms, such as diatoms and radiolaria, secrete skeletal structures made of silica. Silica is deposited in many plant tissues.[11]
History[edit]
Owing to the abundance of silicon in the Earth’s crust, natural silicon-based materials have been used for thousands of years. Silicon rock crystals were familiar to various ancient civilizations, such as the predynastic Egyptians who used it for beads and small vases, as well as the ancient Chinese. Glass containing silica was manufactured by the Egyptians since at least 1500 BC, as well as by the ancient Phoenicians. Natural silicate compounds were also used in various types of mortar for construction of early human dwellings.[12]
Discovery[edit]

In 1787, Antoine Lavoisier suspected that silica might be an oxide of a fundamental chemical element,[13] but the chemical affinity of silicon for oxygen is high enough that he had no means to reduce the oxide and isolate the element.[14] After an attempt to isolate silicon in 1808, Sir Humphry Davy proposed the name «silicium» for silicon, from the Latin silex, silicis for flint, and adding the «-ium» ending because he believed it to be a metal.[15] Most other languages use transliterated forms of Davy’s name, sometimes adapted to local phonology (e.g. German Silizium, Turkish silisyum, Catalan silici, Armenian Սիլիցիում or Silitzioum). A few others use instead a calque of the Latin root (e.g. Russian кремний, from кремень «flint»; Greek πυρίτιο from πυρ «fire»; Finnish pii from piikivi «flint», Czech křemík from křemen «quartz», «flint»).[16]
Gay-Lussac and Thénard are thought to have prepared impure amorphous silicon in 1811, through the heating of recently isolated potassium metal with silicon tetrafluoride, but they did not purify and characterize the product, nor identify it as a new element.[17] Silicon was given its present name in 1817 by Scottish chemist Thomas Thomson. He retained part of Davy’s name but added «-on» because he believed that silicon was a nonmetal similar to boron and carbon.[18] In 1824, Jöns Jacob Berzelius prepared amorphous silicon using approximately the same method as Gay-Lussac (reducing potassium fluorosilicate with molten potassium metal), but purifying the product to a brown powder by repeatedly washing it.[19] As a result, he is usually given credit for the element’s discovery.[20][21] The same year, Berzelius became the first to prepare silicon tetrachloride; silicon tetrafluoride had already been prepared long before in 1771 by Carl Wilhelm Scheele by dissolving silica in hydrofluoric acid.[14] In 1823 for the first time Jacob Berzelius discovered silicon tetrachloride (SiCl4).[22] In 1846 Von Ebelman’s had synthesized Tetraethyl orthosilicate (Si(OC2H5)4).[23][22]
Silicon in its more common crystalline form was not prepared until 31 years later, by Deville.[24][25] By electrolyzing a mixture of sodium chloride and aluminium chloride containing approximately 10% silicon, he was able to obtain a slightly impure allotrope of silicon in 1854.[26] Later, more cost-effective methods have been developed to isolate several allotrope forms, the most recent being silicene in 2010.[27][28] Meanwhile, research on the chemistry of silicon continued; Friedrich Wöhler discovered the first volatile hydrides of silicon, synthesising trichlorosilane in 1857 and silane itself in 1858, but a detailed investigation of the silanes was only carried out in the early 20th century by Alfred Stock, despite early speculation on the matter dating as far back as the beginnings of synthetic organic chemistry in the 1830s.[29][30] Similarly, the first organosilicon compound, tetraethylsilane, was synthesised by Charles Friedel and James Crafts in 1863, but detailed characterisation of organosilicon chemistry was only done in the early 20th century by Frederic Kipping.[14]
Starting in the 1920s, the work of William Lawrence Bragg on X-ray crystallography elucidated the compositions of the silicates, which had previously been known from analytical chemistry but had not yet been understood, together with Linus Pauling’s development of crystal chemistry and Victor Goldschmidt’s development of geochemistry. The middle of the 20th century saw the development of the chemistry and industrial use of siloxanes and the growing use of silicone polymers, elastomers, and resins. In the late 20th century, the complexity of the crystal chemistry of silicides was mapped, along with the solid-state physics of doped semiconductors.[14]
Silicon semiconductors[edit]
The first semiconductor devices did not use silicon, but used galena, including German physicist Ferdinand Braun’s crystal detector in 1874 and Indian physicist Jagadish Chandra Bose’s radio crystal detector in 1901.[31][32] The first silicon semiconductor device was a silicon radio crystal detector, developed by American engineer Greenleaf Whittier Pickard in 1906.[32]
In 1940, Russell Ohl discovered the p–n junction and photovoltaic effects in silicon. In 1941, techniques for producing high-purity germanium and silicon crystals were developed for radar microwave detector crystals during World War II.[31] In 1947, physicist William Shockley theorized a field-effect amplifier made from germanium and silicon, but he failed to build a working device, before eventually working with germanium instead. The first working transistor was a point-contact transistor built by John Bardeen and Walter Brattain later that year while working under Shockley.[33] In 1954, physical chemist Morris Tanenbaum fabricated the first silicon junction transistor at Bell Labs.[34] In 1955, Carl Frosch and Lincoln Derick at Bell Labs accidentally discovered that silicon dioxide (SiO
2) could be grown on silicon,[35] and they later proposed this could mask silicon surfaces during diffusion processes in 1958.[36]
Silicon Age[edit]
The «Silicon Age» refers to the late 20th century to early 21st century.[37][38][39] This is due to silicon being the dominant material of the Silicon Age (also known as the Digital Age or Information Age), similar to how the Stone Age, Bronze Age and Iron Age were defined by the dominant materials during their respective ages of civilization.[37]
Because silicon is an important element in high-technology semiconductor devices, many places in the world bear its name. For example, Santa Clara Valley in California acquired the nickname Silicon Valley, as the element is the base material in the semiconductor industry there. Since then, many other places have been dubbed similarly, including Silicon Wadi in Israel, Silicon Forest in Oregon, Silicon Hills in Austin, Texas, Silicon Slopes in Salt Lake City, Utah, Silicon Saxony in Germany, Silicon Valley in India, Silicon Border in Mexicali, Mexico, Silicon Fen in Cambridge, England, Silicon Roundabout in London, Silicon Glen in Scotland, Silicon Gorge in Bristol, England, Silicon Alley in New York, New York and Silicon Beach in Los Angeles, California.[40]
Characteristics[edit]
Physical and atomic[edit]

A silicon atom has fourteen electrons. In the ground state, they are arranged in the electron configuration [Ne]3s23p2. Of these, four are valence electrons, occupying the 3s orbital and two of the 3p orbitals. Like the other members of its group, the lighter carbon and the heavier germanium, tin, and lead, it has the same number of valence electrons as valence orbitals: hence, it can complete its octet and obtain the stable noble gas configuration of argon by forming sp3 hybrid orbitals, forming tetrahedral SiX
4 derivatives where the central silicon atom shares an electron pair with each of the four atoms it is bonded to.[42] The first four ionisation energies of silicon are 786.3, 1576.5, 3228.3, and 4354.4 kJ/mol respectively; these figures are high enough to preclude the possibility of simple cationic chemistry for the element. Following periodic trends, its single-bond covalent radius of 117.6 pm is intermediate between those of carbon (77.2 pm) and germanium (122.3 pm). The hexacoordinate ionic radius of silicon may be considered to be 40 pm, although this must be taken as a purely notional figure given the lack of a simple Si4+
cation in reality.[43]
Electrical[edit]
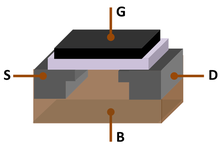
At standard temperature and pressure, silicon is a shiny semiconductor with a bluish-grey metallic lustre; as typical for semiconductors, its resistivity drops as temperature rises. This arises because silicon has a small energy gap (band gap) between its highest occupied energy levels (the valence band) and the lowest unoccupied ones (the conduction band). The Fermi level is about halfway between the valence and conduction bands and is the energy at which a state is as likely to be occupied by an electron as not. Hence pure silicon is effectively an insulator at room temperature. However, doping silicon with a pnictogen such as phosphorus, arsenic, or antimony introduces one extra electron per dopant and these may then be excited into the conduction band either thermally or photolytically, creating an n-type semiconductor. Similarly, doping silicon with a group 13 element such as boron, aluminium, or gallium results in the introduction of acceptor levels that trap electrons that may be excited from the filled valence band, creating a p-type semiconductor.[44] Joining n-type silicon to p-type silicon creates a p–n junction with a common Fermi level; electrons flow from n to p, while holes flow from p to n, creating a voltage drop. This p–n junction thus acts as a diode that can rectify alternating current that allows current to pass more easily one way than the other. A transistor is an n–p–n junction, with a thin layer of weakly p-type silicon between two n-type regions. Biasing the emitter through a small forward voltage and the collector through a large reverse voltage allows the transistor to act as a triode amplifier.[44]
Crystal structure[edit]
Silicon crystallises in a giant covalent structure at standard conditions, specifically in a diamond cubic lattice (space group 227). It thus has a high melting point of 1414 °C, as a lot of energy is required to break the strong covalent bonds and melt the solid. Upon melting silicon contracts as the long-range tetrahedral network of bonds breaks up and the voids in that network are filled in, similar to water ice when hydrogen bonds are broken upon melting. It does not have any thermodynamically stable allotropes at standard pressure, but several other crystal structures are known at higher pressures. The general trend is one of increasing coordination number with pressure, culminating in a hexagonal close-packed allotrope at about 40 gigapascals known as Si–VII (the standard modification being Si–I). An allotrope called BC8 (or bc8), having a body-centred cubic lattice with eight atoms per primitive unit cell (space group 206), can be created at high pressure and remains metastable at low pressure. Its properties have been studied in detail.[45]
Silicon boils at 3265 °C: this, while high, is still lower than the temperature at which its lighter congener carbon sublimes (3642 °C) and silicon similarly has a lower heat of vaporisation than carbon, consistent with the fact that the Si–Si bond is weaker than the C–C bond.[44]
It is also possible to construct silicene layers analogous to graphene.[27][28]
Isotopes[edit]
Naturally occurring silicon is composed of three stable isotopes, 28Si (92.23%), 29Si (4.67%), and 30Si (3.10%).[46] Out of these, only 29Si is of use in NMR and EPR spectroscopy,[47] as it is the only one with a nuclear spin (I =1/2).[29] All three are produced in Type Ia supernovae[48][49] through the oxygen-burning process, with 28Si being made as part of the alpha process and hence the most abundant. The fusion of 28Si with alpha particles by photodisintegration rearrangement in stars is known as the silicon-burning process; it is the last stage of stellar nucleosynthesis before the rapid collapse and violent explosion of the star in question in a type II supernova.[50]
Twenty radioisotopes have been characterized, the two stablest being 32Si with a half-life of about 150 years, and 31Si with a half-life of 2.62 hours.[46] All the remaining radioactive isotopes have half-lives that are less than seven seconds, and the majority of these have half-lives that are less than one tenth of a second.[46] Silicon has one known nuclear isomer, 34mSi, with a half-life less than 210 nanoseconds.[46] 32Si undergoes low-energy beta decay to 32P and then stable 32S. 31Si may be produced by the neutron activation of natural silicon and is thus useful for quantitative analysis; it can be easily detected by its characteristic beta decay to stable 31P, in which the emitted electron carries up to 1.48 MeV of energy.[29]
The known isotopes of silicon range in mass number from 22 to 44.[46] The most common decay mode of the isotopes with mass numbers lower than the three stable isotopes is inverse beta decay, primarily forming aluminium isotopes (13 protons) as decay products.[46] The most common decay mode for the heavier unstable isotopes is beta decay, primarily forming phosphorus isotopes (15 protons) as decay products.[46]
Silicon can enter the oceans through groundwater and riverine transport. Large fluxes of groundwater input have an isotopic composition which is distinct from riverine silicon inputs. Isotopic variations in groundwater and riverine transports contribute to variations in oceanic 30Si values. Currently, there are substantial differences in the isotopic values of deep water in the world’s ocean basins. Between the Atlantic and Pacific oceans, there is a deep water 30Si gradient of greater than 0.3 parts per thousand. 30Si is most commonly associated with productivity in the oceans.[51]
Chemistry and compounds[edit]
| X = | C | Si | H | F | Cl | Br | I | O– | N< |
|---|---|---|---|---|---|---|---|---|---|
| C–X | 368 | 360 | 435 | 453 | 351 | 293 | 216 | ~360 | ~305 |
| Si–X | 360 | 340 | 393 | 565 | 381 | 310 | 234 | 452 | 322 |
Crystalline bulk silicon is rather inert, but becomes more reactive at high temperatures. Like its neighbour aluminium, silicon forms a thin, continuous surface layer of silicon dioxide (SiO
2) that protects the metal from oxidation. Thus silicon does not measurably react with the air below 900 °C, but formation of the vitreous dioxide rapidly increases between 950 °C and 1160 °C and when 1400 °C is reached, atmospheric nitrogen also reacts to give the nitrides SiN and Si
3N
4. Silicon reacts with gaseous sulfur at 600 °C and gaseous phosphorus at 1000 °C. This oxide layer nevertheless does not prevent reaction with the halogens; fluorine attacks silicon vigorously at room temperature, chlorine does so at about 300 °C, and bromine and iodine at about 500 °C. Silicon does not react with most aqueous acids, but is oxidised and complexed by hydrofluoric acid mixtures containing either chlorine or nitric acid to form hexafluorosilicates. It readily dissolves in hot aqueous alkali to form silicates.[52] At high temperatures, silicon also reacts with alkyl halides; this reaction may be catalysed by copper to directly synthesise organosilicon chlorides as precursors to silicone polymers. Upon melting, silicon becomes extremely reactive, alloying with most metals to form silicides, and reducing most metal oxides because the heat of formation of silicon dioxide is so large. In fact, molten silicon reacts virtually with every known kind of crucible material (except its own oxide, SiO
2).[53]: 13 This happens due to silicon’s high binding forces for the light elements and to its high dissolving power for most elements.[53]: 13 As a result, containers for liquid silicon must be made of refractory, unreactive materials such as zirconium dioxide or group 4, 5, and 6 borides.[44][54]
Tetrahedral coordination is a major structural motif in silicon chemistry just as it is for carbon chemistry. However, the 3p subshell is rather more diffuse than the 2p subshell and does not hybridise so well with the 3s subshell. As a result, the chemistry of silicon and its heavier congeners shows significant differences from that of carbon,[55] and thus octahedral coordination is also significant.[44] For example, the electronegativity of silicon (1.90) is much less than that of carbon (2.55), because the valence electrons of silicon are further from the nucleus than those of carbon and hence experience smaller electrostatic forces of attraction from the nucleus. The poor overlap of 3p orbitals also results in a much lower tendency toward catenation (formation of Si–Si bonds) for silicon than for carbon, due to the concomitant weakening of the Si–Si bond compared to the C–C bond:[56] the average Si–Si bond energy is approximately 226 kJ/mol, compared to a value of 356 kJ/mol for the C–C bond.[57] This results in multiply bonded silicon compounds generally being much less stable than their carbon counterparts, an example of the double bond rule. On the other hand, the presence of radial nodes in the 3p orbitals of silicon suggests the possibility of hypervalence, as seen in five and six-coordinate derivatives of silicon such as SiX−
5 and SiF2−
6.[58][56] Lastly, because of the increasing energy gap between the valence s and p orbitals as the group is descended, the divalent state grows in importance from carbon to lead, so that a few unstable divalent compounds are known for silicon; this lowering of the main oxidation state, in tandem with increasing atomic radii, results in an increase of metallic character down the group. Silicon already shows some incipient metallic behavior, particularly in the behavior of its oxide compounds and its reaction with acids as well as bases (though this takes some effort), and is hence often referred to as a metalloid rather than a nonmetal.[56] However, metallicity does not become clear in group 14 until germanium and dominant until tin, with the growing importance of the lower +2 oxidation state.[14]
Silicon shows clear differences from carbon. For example, organic chemistry has very few analogies with silicon chemistry, while silicate minerals have a structural complexity unseen in oxocarbons.[59] Silicon tends to resemble germanium far more than it does carbon, and this resemblance is enhanced by the d-block contraction, resulting in the size of the germanium atom being much closer to that of the silicon atom than periodic trends would predict.[60] Nevertheless, there are still some differences because of the growing importance of the divalent state in germanium compared to silicon, which result in germanium being significantly more metallic than silicon. Additionally, the lower Ge–O bond strength compared to the Si–O bond strength results in the absence of «germanone» polymers that would be analogous to silicone polymers.[57]
Silicides[edit]
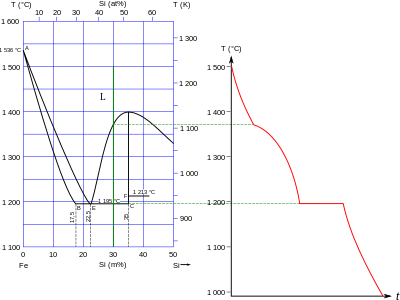
Phase diagram of the Fe–Si system
Many metal silicides are known, most of which have formulae that cannot be explained through simple appeals to valence: their bonding ranges from metallic to ionic and covalent. Some known stoichiometries are M
6Si, M
5Si, M
4Si, M
15Si
4, M
3Si, M
5Si
2, M
2Si, M
5Si
3, M
3Si
2, MSi, M
2Si
3, MSi
2, MSi
3, and MSi
6. They are structurally more similar to the borides than the carbides, in keeping with the diagonal relationship between boron and silicon, although the larger size of silicon than boron means that exact structural analogies are few and far between. The heats of formation of the silicides are usually similar to those of the borides and carbides of the same elements, but they usually melt at lower temperatures.[61] Silicides are known for all stable elements in groups 1–10, with the exception of beryllium: in particular, uranium and the transition metals of groups 4–10 show the widest range of stoichiometries. Except for copper, the metals in groups 11–15 do not form silicides. Instead, most form eutectic mixtures, although the heaviest post-transition metals mercury, thallium, lead, and bismuth are completely immiscible with liquid silicon.[44]
Usually, silicides are prepared by direct reaction of the elements. For example, the alkali metals and alkaline earth metals react with silicon or silicon oxide to give silicides. Nevertheless, even with these highly electropositive elements true silicon anions are not obtainable, and most of these compounds are semiconductors. For example, the alkali metal silicides (M+
)
4(Si4−
4) contain pyramidal tricoordinate silicon in the Si4−
4 anion, isoelectronic with white phosphorus, P
4.[44][62] Metal-rich silicides tend to have isolated silicon atoms (e. g. Cu
5Si); with increasing silicon content, catenation increases, resulting in isolated clusters of two (e. g. U
3Si
2) or four silicon atoms (e. g. [K+
]
4[Si
4]4−
) at first, followed by chains (e. g. CaSi), layers (e. g. CaSi
2), or three-dimensional networks of silicon atoms spanning space (e. g. α-ThSi
2) as the silicon content rises even higher.[44]
The silicides of the group 1 and 2 metals usually are more reactive than the transition metal silicides. The latter usually do not react with aqueous reagents, except for hydrofluoric acid; however, they do react with much more aggressive reagents such as liquid potassium hydroxide, or gaseous fluorine or chlorine when red-hot. The pre-transition metal silicides instead readily react with water and aqueous acids, usually producing hydrogen or silanes:[44]
- Na
2Si + 3 H2O → Na
2SiO
3 + 3 H
2 - Mg
2Si + 2 H
2SO
4 → 2 MgSO
4 + SiH
4
Products often vary with the stoichiometry of the silicide reactant. For example, Ca
2Si is polar and non-conducting and has the anti-PbCl
2 structure with single isolated silicon atoms, and reacts with water to produce calcium hydroxide, hydrated silicon dioxide, and hydrogen gas. CaSi with its zigzag chains of silicon atoms instead reacts to give silanes and polymeric SiH
2, while CaSi
2 with its puckered layers of silicon atoms does not react with water, but will react with dilute hydrochloric acid: the product is a yellow polymeric solid with stoichiometry Si
2H
2O.[44]
Silanes[edit]
Speculation on silicon hydride chemistry started in the 1830s, contemporary with the development of synthetic organic chemistry. Silane itself, as well as trichlorosilane, were first synthesised by Friedrich Wöhler and Heinrich Buff in 1857 by reacting aluminium–silicon alloys with hydrochloric acid, and characterised as SiH
4 and SiHCl
3 by Charles Friedel and Albert Ladenburg in 1867. Disilane (Si
2H
6) followed in 1902, when it was first made by Henri Moissan and Samuel Smiles by the protonolysis of magnesium silicides. Further investigation had to wait until 1916 because of the great reactivity and thermal instability of the silanes; it was then that Alfred Stock began to study silicon hydrides in earnest with new greaseless vacuum techniques, as they were found as contaminants of his focus, the boron hydrides. The names silanes and boranes are his, based on analogy with the alkanes.[29][63][64] The Moissan and Smiles method of preparation of silanes and silane derivatives via protonolysis of metal silicides is still used, although the yield is lowered by the hydrolysis of the products that occurs simultaneously, so that the preferred route today is to treat substituted silanes with hydride reducing agents such as lithium aluminium hydride in etheric solutions at low temperatures. Direct reaction of HX or RX with silicon, possibly with a catalyst such as copper, is also a viable method of producing substituted silanes.[29]
The silanes comprise a homologous series of silicon hydrides with a general formula of Si
nH
2n + 2. They are all strong reducing agents. Unbranched and branched chains are known up to n=8, and the cycles Si
5H
10 and Si
6H
12 are also known. The first two, silane and disilane, are colourless gases; the heavier members of the series are volatile liquids. All silanes are very reactive and catch fire or explode spontaneously in air. They become less thermally stable with room temperature, so that only silane is indefinitely stable at room temperature, although disilane does not decompose very quickly (only 2.5% of a sample decomposes after the passage of eight months).[29] They decompose to form polymeric polysilicon hydride and hydrogen gas.[65][66] As expected from the difference in atomic weight, the silanes are less volatile than the corresponding alkanes and boranes, but more so than the corresponding germanes. They are much more reactive than the corresponding alkanes, because of the larger radius of silicon compared to carbon facilitating nucleophilic attack at the silicon, the greater polarity of the Si–H bond compared to the C–H bond, and the ability of silicon to expand its octet and hence form adducts and lower the reaction’s activation energy.[29]
Silane pyrolysis gives polymeric species and finally elemental silicon and hydrogen; indeed ultrapure silicon is commercially produced by the pyrolysis of silane. While the thermal decomposition of alkanes starts by the breaking of a C–H or C–C bond and the formation of radical intermediates, polysilanes decompose by eliminating silylenes :SiH
2 or :SiHR, as the activation energy of this process (~210 kJ/mol) is much less than the Si–Si and Si–H bond energies. While pure silanes do not react with pure water or dilute acids, traces of alkali catalyse immediate hydrolysis to hydrated silicon dioxide. If the reaction is carried out in methanol, controlled solvolysis results in the products SiH
2(OMe)
2, SiH(OMe)
3, and Si(OMe)
4. The Si–H bond also adds to alkenes, a reaction which proceeds slowly and speeds up with increasing substitution of the silane involved. At 450 °C, silane participates in an addition reaction with acetone, as well as a ring-opening reaction with ethylene oxide. Direct reaction of the silanes with chlorine or bromine results in explosions at room temperature, but the reaction of silane with bromine at −80 °C is controlled and yields bromosilane and dibromosilane. The monohalosilanes may be formed by reacting silane with the appropriate hydrogen halide with an Al
2X
6 catalyst, or by reacting silane with a solid silver halide in a heated flow reactor:[29]
- SiH
4 + 2 AgCl 260 °C→ SiH
3Cl + HCl + 2 Ag
Among the derivatives of silane, iodosilane (SiH
3I) and potassium silanide (KSiH
3) are very useful synthetic intermediates in the production of more complicated silicon-containing compounds: the latter is a colourless crystalline ionic solid containing K+ cations and SiH−
3 anions in the NaCl structure, and is made by the reduction of silane by potassium metal.[67] Additionally, the reactive hypervalent species SiH−
5 is also known.[29] With suitable organic substituents it is possible to produce stable polysilanes: they have surprisingly high electric conductivities, arising from sigma delocalisation of the electrons in the chain.[68]
Halides[edit]
Silicon and silicon carbide readily react with all four stable halogens, forming the colourless, reactive, and volatile silicon tetrahalides[69] Silicon tetrafluoride also may be made by fluorinating the other silicon halides, and is produced by the attack of hydrofluoric acid on glass.[70] Heating two different tetrahalides together also produces a random mixture of mixed halides, which may also be produced by halogen exchange reactions. The melting and boiling points of these species usually rise with increasing atomic weight, though there are many exceptions: for example, the melting and boiling points drop as one passes from SiFBr
3 through SiFClBr
2 to SiFCl
2Br. The shift from the hypoelectronic elements in Group 13 and earlier to the Group 14 elements is illustrated by the change from an infinite ionic structure in aluminium fluoride to a lattice of simple covalent silicon tetrafluoride molecules, as dictated by the lower electronegativity of aluminium than silicon, the stoichiometry (the +4 oxidation state being too high for true ionicity), and the smaller size of the silicon atom compared to the aluminium atom.[69]
Silicon tetrachloride is manufactured on a huge scale as a precursor to the production of pure silicon, silicon dioxide, and some silicon esters.[69] The silicon tetrahalides hydrolyse readily in water, unlike the carbon tetrahalides, again because of the larger size of the silicon atom rendering it more open to nucleophilic attack and the ability of the silicon atom to expand its octet which carbon lacks.[70] The reaction of silicon tetrafluoride with excess hydrofluoric acid produces the octahedral hexafluorosilicate anion SiF2−
6.[70]
Analogous to the silanes, halopolysilanes Si
nX
2n + 2 also are known. While catenation in carbon compounds is maximised in the hydrogen compounds rather than the halides, the opposite is true for silicon, so that the halopolysilanes are known up to at least Si
14F
30, Si
6Cl
14, and Si
4Br
10. A suggested explanation for this phenomenon is the compensation for the electron loss of silicon to the more electronegative halogen atoms by pi backbonding from the filled pπ orbitals on the halogen atoms to the empty dπ orbitals on silicon: this is similar to the situation of carbon monoxide in metal carbonyl complexes and explains their stability. These halopolysilanes may be produced by comproportionation of silicon tetrahalides with elemental silicon, or by condensation of lighter halopolysilanes (trimethylammonium being a useful catalyst for this reaction).[69]
Silica[edit]
Silicon dioxide (SiO
2), also known as silica, is one of the best-studied compounds, second only to water. Twelve different crystal modifications of silica are known, the most common being α-quartz, a major constituent of many rocks such as granite and sandstone. It also is known to occur in a pure form as rock crystal; impure forms are known as rose quartz, smoky quartz, morion, amethyst, and citrine. Some poorly crystalline forms of quartz are also known, such as chalcedony, chrysoprase, carnelian, agate, onyx, jasper, heliotrope, and flint. Other modifications of silicon dioxide are known in some other minerals such as tridymite and cristobalite, as well as the much less common coesite and stishovite. Biologically generated forms are also known as kieselguhr and diatomaceous earth. Vitreous silicon dioxide is known as tektites, and obsidian, and rarely as lechatelierite. Some synthetic forms are known as keatite. Opals are composed of complicated crystalline aggregates of partially hydrated silicon dioxide.[71]
-

Quartz
-

Agate
-

Tridymite
-

Cristobalite
-

Coesite





Most crystalline forms of silica are made of infinite arrangements of SiO tetrahedra (with Si at the center) connected at their corners, with each oxygen atom linked to two silicon atoms. In the thermodynamically stable room-temperature form, α-quartz, these tetrahedra are linked in intertwined helical chains with two different Si–O distances (159.7 and 161.7 pm) with a Si–O–Si angle of 144°. These helices can be either left- or right-handed, so that individual α-quartz crystals are optically active. At 537 °C, this transforms quickly and reversibly into the similar β-quartz, with a change of the Si–O–Si angle to 155° but a retention of handedness. Further heating to 867 °C results in another reversible phase transition to β-tridymite, in which some Si–O bonds are broken to allow for the arrangement of the SiO tetrahedra into a more open and less dense hexagonal structure. This transition is slow and hence tridymite occurs as a metastable mineral even below this transition temperature; when cooled to about 120 °C it quickly and reversibly transforms by slight displacements of individual silicon and oxygen atoms to α-tridymite, similarly to the transition from α-quartz to β-quartz. β-tridymite slowly transforms to cubic β-cristobalite at about 1470 °C, which once again exists metastably below this transition temperature and transforms at 200–280 °C to α-cristobalite via small atomic displacements. β-cristobalite melts at 1713 °C; the freezing of silica from the melt is quite slow and vitrification, or the formation of a glass, is likely to occur instead. In vitreous silica, the SiO tetrahedra remain corner-connected, but the symmetry and periodicity of the crystalline forms are lost. Because of the slow conversions between these three forms, it is possible upon rapid heating to melt β-quartz (1550 °C) or β-tridymite (1703 °C). Silica boils at approximately 2800 °C. Other high-pressure forms of silica are known, such as coesite and stishovite: these are known in nature, formed under the shock pressure of a meteorite impact and then rapidly quenched to preserve the crystal structure. Similar melting and cooling of silica occurs following lightning strikes, forming glassy lechatelierite. W-silica is an unstable low-density form involving SiO tetrahedra sharing opposite edges instead of corners, forming parallel chains similarly to silicon disulfide (SiS
2) and silicon diselenide (SiSe
2): it quickly returns to forming amorphous silica with heat or traces of water[72]

Condensed polysilicic acid
Silica is rather inert chemically. It is not attacked by any acids other than hydrofluoric acid. However, it slowly dissolves in hot concentrated alkalis, and does so rather quickly in fused metal hydroxides or carbonates, to give metal silicates. Among the elements, it is attacked only by fluorine at room temperature to form silicon tetrafluoride: hydrogen and carbon also react, but require temperatures over 1000 °C to do so. Silica nevertheless reacts with many metal and metalloid oxides to form a wide variety of compounds important in the glass and ceramic industries above all, but also have many other uses: for example, sodium silicate is often used in detergents due to its buffering, saponifying, and emulsifying properties[72]
Silicic acids[edit]
Adding water to silica drops its melting point by around 800 °C due to the breaking of the structure by replacing Si–O–Si linkages with terminating Si–OH groups. Increasing water concentration results in the formation of hydrated silica gels and colloidal silica dispersions. Many hydrates and silicic acids exist in the most dilute of aqueous solutions, but these are rather insoluble and quickly precipitate and condense and cross-link to form various polysilicic acids of variable combinations following the formula [SiO
x(OH)
4−2x]
n, similar to the behaviour of boron, aluminium, and iron, among other elements. Hence, although some simple silicic acids have been identified in dilute solutions, such as orthosilicic acid Si(OH)
4 and metasilicic acid SiO(OH)
2, none of these are likely to exist in the solid state.[72]
Silicate minerals[edit]
| CN 4 | LiI (59) |
BeII (27) | AlIII (39) | SiIV (26) | |
|---|---|---|---|---|---|
| CN 6 | NaI (102) | MgII (72) | AlIII (54) | TiIV (61) | FeII (78) |
| CN 8 | KI (151) | CaII (112) | |||
| CN 12 | KI (164) |
About 95% of the Earth’s crustal rocks are made of silica or silicate and aluminosilicate minerals, as reflected in oxygen, silicon, and aluminium being the three most common elements in the crust (in that order).[73] Measured by mass, silicon makes up 27.7% of the Earth’s crust.[74] Pure silicon crystals are very rarely found in nature, but notable exceptions are crystals as large as to 0.3 mm across found during sampling gases from the Kudriavy volcano on Iturup, one of the Kuril Islands.[75][76]
Silicate and aluminosilicate minerals have many different structures and varying stoichiometry, but they may be classified following some general principles. Tetrahedral SiO units are common to almost all these compounds, either as discrete structures, or combined into larger units by the sharing of corner oxygen atoms. These may be divided into neso-silicates (discrete SiO units) sharing no oxygen atoms, soro-silicates (discrete Si units) sharing one, cyclo-silicates (closed ring structures) and ino-silicates (continuous chain or ribbon structures) both sharing two, phyllo-silicates (continuous sheets) sharing three, and tecto-silicates (continuous three-dimensional frameworks) sharing four. The lattice of oxygen atoms that results is usually close-packed, or close to it, with the charge being balanced by other cations in various different polyhedral sites according to size.[77]
The orthosilicates MII
2SiO
4 (M = Be, Mg, Mn, Fe, Zn) and ZrSiO
4 are neso-silicates. Be
2SiO
4 (phenacite) is unusual as both BeII and SiIV occupy tetrahedral four-coordinated sites; the other divalent cations instead occupy six-coordinated octahedral sites and often isomorphously replace each other as in olivine, (Mg,Fe,Mn)
2SiO
4. Zircon, ZrSiO
4, demands eight-coordination of the ZrIV cations due to stoichiometry and because of their larger ionic radius (84 pm). Also significant are the garnets, [MII
3MIII
2(SiO
4)
3], in which the divalent cations (e.g. Ca, Mg, Fe) are eight-coordinated and the trivalent ones are six-coordinated (e.g. Al, Cr, Fe). Regular coordination is not always present: for example, it is not found in Ca
2SiO
4, which mixes six- and eight-coordinate sites for CaII. Soro-silicates, involving discrete double or triple tetrahedral units, are quite rare: metasilicates involving cyclic «[(SiOn
3)]2n−» units of corner-abutting tetrahedra forming a polygonal ring are also known.[73]
Chain metasilicates, {SiO2−
3}
∞, form by corner-sharing of an indefinite chain of linked SiO tetrahedra. Many differences arise due to the differing repeat distances of conformation across the line of tetrahedra. A repeat distance of two is most common, as in most pyroxene minerals, but repeat distances of one, three, four, five, six, seven, nine, and twelve are also known. These chains may then link across each other to form double chains and ribbons, as in the asbestos minerals, involving repeated chains of cyclic tetrahedron rings.[73]

A typical zeolite structure
Layer silicates, such as the clay minerals and the micas, are very common, and often are formed by horizontal cross-linking of metasilicate chains or planar condensation of smaller units. An example is kaolinite [Al
2(OH)
4Si
2O
5]; in many of these minerals cation and anion replacement is common, so that for example tetrahedral SiIV may be replaced by AlIII, octahedral AlIII by MgII, and OH−
by F−
. Three-dimensional framework aluminosilicates are structurally very complex; they may be conceived of as starting from the SiO
2 structure, but having replaced up to one-half of the SiIV atoms with AlIII, they require more cations to be included in the structure to balance charge. Examples include feldspars (the most abundant minerals on the Earth), zeolites, and ultramarines. Many feldspars can be thought of as forming part of the ternary system NaAlSi
3O
8–KAlSi
3O
8–CaAl
2Si
2O
8. Their lattice is destroyed by high pressure prompting AlIII to undergo six-coordination rather than four-coordination, and this reaction destroying feldspars may be a reason for the Mohorovičić discontinuity, which would imply that the crust and mantle have the same chemical composition, but different lattices, although this is not a universally held view. Zeolites have many polyhedral cavities in their frameworks (truncated cuboctahedra being most common, but other polyhedra also are known as zeolite cavities), allowing them to include loosely bound molecules such as water in their structure. Ultramarines alternate silicon and aluminium atoms and include a variety of other anions such as Cl−, SO2−
4, and S2−
2, but are otherwise similar to the feldspars.[73]
Other inorganic compounds[edit]
Silicon disulfide (SiS
2) is formed by burning silicon in gaseous sulfur at 100 °C; sublimation of the resulting compound in nitrogen results in white, flexible long fibers reminiscent of asbestos with a structure similar to W-silica. This melts at 1090 °C and sublimes at 1250 °C; at high temperature and pressure this transforms to a crystal structure analogous to cristobalite. However, SiS
2 lacks the variety of structures of SiO
2, and quickly hydrolyses to silica and hydrogen sulfide. It is also ammonolysed quickly and completely by liquid ammonia as follows to form an imide:[60]
- SiS
2 + 4 NH
3 → Si(NH)
2 + 2 NH
4SH
It reacts with the sulfides of sodium, magnesium, aluminium, and iron to form metal thiosilicates: reaction with ethanol results in tetraethylsilicate Si(OEt)
4 and hydrogen sulfide. Ethylsilicate is useful as its controlled hydrolysis produces adhesive or film-like forms of silica. Reacting hydrogen sulfide with silicon tetrahalides yields silicon thiohalides such as S(SiCl)
3, cyclic Cl
2Si(μ-S)
2SiCl
2, and crystalline (SiSCl
2)
4. Despite the double bond rule, stable organosilanethiones RR’Si=S have been made thanks to the stabilising mechanism of intermolecular coordination via an amine group.[78]
Silicon nitride, Si
3N
4, may be formed by directly reacting silicon with nitrogen above 1300 °C, but a more economical means of production is by heating silica and coke in a stream of nitrogen and hydrogen gas at 1500 °C. It would make a promising ceramic if not for the difficulty of working with and sintering it: chemically, it is near-totally inert, and even above 1000 °C it keeps its strength, shape, and continues to be resistant to wear and corrosion. It is very hard (9 on the Mohs hardness scale), dissociates only at 1900 °C at 1 atm, and is quite dense (density 3.185 g/cm3), because of its compact structure similar to that of phenacite (Be
2SiO
4). A similar refractory material is Si
2N
2O, formed by heating silicon and silica at 1450 °C in an argon stream containing 5% nitrogen gas, involving 4-coordinate silicon and 3-coordinate nitrogen alternating in puckered hexagonal tilings interlinked by non-linear Si–O–Si linkages to each other.[78]
Reacting silyl halides with ammonia or alkylammonia derivatives in the gaseous phase or in ethanolic solution produces various volatile silylamides, which are silicon analogues of the amines:[78]
- 3 SiH
3Cl + 4 NH
3 → N(SiH
3)
3 + 3 NH
4Cl - SiH
3Br + 2 Me
2NH → SiH
3NMe
2 + Me
2NH
2Br - 4 SiH
3I + 5 N
2H
4 → (SiH
3)
2NN(SiH
3)
2 + 4 N
2H
5I
Many such compounds have been prepared, the only known restriction being that the nitrogen is always tertiary, and species containing the SiH–NH group are unstable at room temperature. The stoichiometry around the nitrogen atom in compounds such as N(SiH
3)
3 is planar. Similarly, trisilylamines are weaker as ligands than their carbon analogues, the tertiary amines, although substitution of some SiH
3 groups by CH
3 groups mitigates this weakness. For example, N(SiH
3)
3 does not form an adduct with BH
3 at all, while MeN(SiH
3)
2 and Me
2NSiH
3 form adducts at low temperatures that decompose upon warming. Some silicon analogues of imines, with a Si=N double bond, are known: the first found was But2Si=N–SiBut3, which was discovered in 1986.[78]

Silicon carbide (SiC) was first made by Edward Goodrich Acheson in 1891, who named it carborundum to reference its intermediate hardness and abrasive power between diamond (an allotrope of carbon) and corundum (aluminium oxide). He soon founded a company to manufacture it, and today about one million tonnes are produced each year.[79] Silicon carbide exists in about 250 crystalline forms.[80] The polymorphism of SiC is characterized by a large family of similar crystalline structures called polytypes. They are variations of the same chemical compound that are identical in two dimensions and differ in the third. Thus they can be viewed as layers stacked in a certain sequence.[81] It is made industrially by reduction of quartz sand with excess coke or anthracite at 2000–2500 °C in an electric furnace:[79]
- SiO
2 + 2 C → Si + 2 CO - Si + C → SiC
It is the most thermally stable binary silicon compound, only decomposing through loss of silicon starting from around 2700 °C. It is resistant to most aqueous acids, phosphoric acid being an exception. It forms a protective layer of silicon dioxide on the surface and hence only oxidises appreciably in air above 1000 °C; removal of this layer by molten hydroxides or carbonates leads to quick oxidation. Silicon carbide is rapidly attacked by chlorine gas, which forms SiCl
4 and carbon at 100 °C and SiCl
4 and CCl
4 at 1000 °C. It is mostly used as an abrasive and a refractory material, as it is chemically stable and very strong, and it fractures to form a very sharp cutting edge. It is also useful as an intrinsic semiconductor, as well as an extrinsic semiconductor upon being doped.[79] In its diamond-like behavior it serves as an illustration of the chemical similarity between carbon and silicon.[82]
Organosilicon compounds[edit]

A hydrosilylation reaction, in which Si–H is added to an unsaturated substrate
Because the Si–C bond is close in strength to the C–C bond, organosilicon compounds tend to be markedly thermally and chemically stable. For example, tetraphenylsilane (SiPh
4) may be distilled in air even at its boiling point of 428 °C, and so may its substituted derivatives Ph
3SiCl and Ph
2SiCl
2, which boil at 378 °C and 305 °C respectively. Furthermore, since carbon and silicon are chemical congeners, organosilicon chemistry shows some significant similarities with carbon chemistry, for example in the propensity of such compounds for catenation and forming multiple bonds.[82] However, significant differences also arise: since silicon is more electropositive than carbon, bonds to more electronegative elements are generally stronger with silicon than with carbon, and vice versa. Thus the Si–F bond is significantly stronger than even the C–F bond and is one of the strongest single bonds, while the Si–H bond is much weaker than the C–H bond and is readily broken. Furthermore, the ability of silicon to expand its octet is not shared by carbon, and hence some organosilicon reactions have no organic analogues. For example, nucleophilic attack on silicon does not proceed by the SN2 or SN1 processes, but instead goes through a negatively charged true pentacoordinate intermediate and appears like a substitution at a hindered tertiary atom. This works for silicon, unlike for carbon, because the long Si–C bonds reduce the steric hindrance and there are no geometric constraints for nucleophilic attack, unlike for example a C–O σ* antibonding orbital. Nevertheless, despite these differences, the mechanism is still often called «SN2 at silicon» for simplicity.[83]
One of the most useful silicon-containing groups is trimethylsilyl, Me
3Si–. The Si–C bond connecting it to the rest of the molecule is reasonably strong, allowing it to remain while the rest of the molecule undergoes reactions, but is not so strong that it cannot be removed specifically when needed, for example by the fluoride ion, which is a very weak nucleophile for carbon compounds but a very strong one for organosilicon compounds. It may be compared to acidic protons; while trisilylmethyl is removed by hard nucleophiles instead of bases, both removals usually promote elimination. As a general rule, while saturated carbon is best attacked by nucleophiles that are neutral compounds, those based on nonmetals far down on the periodic table (e.g. sulfur, selenium, or iodine), or even both, silicon is best attacked by charged nucleophiles, particularly those involving such highly electronegative nonmetals as oxygen, fluorine, or chlorine. For example, enolates react at the carbon in haloalkanes, but at the oxygen in silyl chlorides; and when trimethylsilyl is removed from an organic molecule using hydroxide as a nucleophile, the product of the reaction is not the silanol as one would expect from using carbon chemistry as an analogy, because the siloxide is strongly nucleophilic and attacks the original molecule to yield the silyl ether hexamethyldisiloxane, (Me
3Si)
2O. Conversely, while the SN2 reaction is mostly unaffected by the presence of a partial positive charge (δ+) at the carbon, the analogous «SN2″ reaction at silicon is so affected. Thus, for example, the silyl triflates are so electrophilic that they react 108 to 109 times faster than silyl chlorides with oxygen-containing nucleophiles. Trimethylsilyl triflate is in particular a very good Lewis acid and is used to convert carbonyl compounds to acetals and silyl enol ethers, reacting them together analogously to the aldol reaction.[83]
Si–C bonds are commonly formed in three ways. In the laboratory, preparation is often carried out in small quantities by reacting tetrachlorosilane (silicon tetrachloride) with organolithium, Grignard, or organoaluminium reagents, or by catalytic addition of Si–H across C=C double bonds. The second route has the drawback of not being applicable to the most important silanes, the methyl and phenyl silanes. Organosilanes are made industrially by directly reacting alkyl or aryl halides with silicon with 10% by weight metallic copper as a catalyst. Standard organic reactions suffice to produce many derivatives; the resulting organosilanes are often significantly more reactive than their carbon congeners, readily undergoing hydrolysis, ammonolysis, alcoholysis, and condensation to form cyclic oligomers or linear polymers.[82]
Silicone polymers[edit]
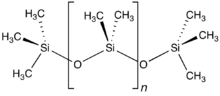
The word «silicone» was first used by Frederic Kipping in 1901. He invented the word to illustrate the similarity of chemical formulae between Ph
2SiO and benzophenone, Ph
2CO, although he also stressed the lack of chemical resemblance due to the polymeric structure of Ph
2SiO, which is not shared by Ph
2CO.[82]
Silicones may be considered analogous to mineral silicates, in which the methyl groups of the silicones correspond to the isoelectronic <O−
of the silicates.[82] They are quite stable to extreme temperatures, oxidation, and water, and have useful dielectric, antistick, and antifoam properties. Furthermore, they are resistant over long periods of time to ultraviolet radiation and weathering, and are inert physiologically. They are fairly unreactive, but do react with concentrated solutions bearing the hydroxide ion and fluorinating agents, and occasionally, may even be used as mild reagents for selective syntheses. For example, (Me
3Si)
2O is valuable for the preparation of derivatives of molybdenum and tungsten oxyhalides, converting a tungsten hexachloride suspension in dichloroethane solution quantitatively to WOCl
4 in under an hour at room temperature, and then to yellow WO
2Cl
2 at 100 °C in light petroleum at a yield of 95% overnight.[84]
Occurrence[edit]

Silicon is the eighth most abundant element in the universe, coming after hydrogen, helium, carbon, nitrogen, oxygen, iron, and neon. These abundances are not replicated well on Earth due to substantial separation of the elements taking place during the formation of the Solar System. Silicon makes up 27.2% of the Earth’s crust by weight, second only to oxygen at 45.5%, with which it always is associated in nature. Further fractionation took place in the formation of the Earth by planetary differentiation: Earth’s core, which makes up 31.5% of the mass of the Earth, has approximate composition Fe
25Ni
2Co
0.1S
3; the mantle makes up 68.1% of the Earth’s mass and is composed mostly of denser oxides and silicates, an example being olivine, (Mg,Fe)
2SiO
4; while the lighter siliceous minerals such as aluminosilicates rise to the surface and form the crust, making up 0.4% of the Earth’s mass.[85][86]
The crystallisation of igneous rocks from magma depends on a number of factors; among them are the chemical composition of the magma, the cooling rate, and some properties of the individual minerals to be formed, such as lattice energy, melting point, and complexity of their crystal structure. As magma is cooled, olivine appears first, followed by pyroxene, amphibole, biotite mica, orthoclase feldspar, muscovite mica, quartz, zeolites, and finally, hydrothermal minerals. This sequence shows a trend toward increasingly complex silicate units with cooling, and the introduction of hydroxide and fluoride anions in addition to oxides. Many metals may substitute for silicon. After these igneous rocks undergo weathering, transport, and deposition, sedimentary rocks like clay, shale, and sandstone are formed. Metamorphism also may occur at high temperatures and pressures, creating an even vaster variety of minerals.[85]
There are four sources for silicon fluxes into the ocean include chemical weathering of continental rocks, river transport, dissolution of continental terrigenous silicates, and through the reaction between submarine basalts and hydrothermal fluid which release dissolved silicon. All four of these fluxes are interconnected in the ocean’s biogeochemical cycle as they all were initially formed from the weathering of Earth’s crust.[87]
Approximately 300–900 megatonnes of Aeolian dust is deposited into the world’s oceans each year. Of that value, 80–240 megatonnes are in the form of particulate silicon. The total amount of particulate silicon deposition into the ocean is still less than the amount of silicon influx into the ocean via riverine transportation.[88] Aeolian inputs of particulate lithogenic silicon into the North Atlantic and Western North Pacific oceans are the result of dust settling on the oceans from the Sahara and Gobi Desert, respectively.[87] Riverine transports are the major source of silicon influx into the ocean in coastal regions, while silicon deposition in the open ocean is greatly influenced by the settling of Aeolian dust.[88]
Production[edit]
Silicon of 96–99% purity is made by reducing quartzite or sand with highly pure coke. The reduction is carried out in an electric arc furnace, with an excess of SiO
2 used to stop silicon carbide (SiC) from accumulating:[29]
- SiO
2 + 2 C → Si + 2 CO - 2 SiC + SiO
2 → 3 Si + 2 CO

This reaction, known as carbothermal reduction of silicon dioxide, usually is conducted in the presence of scrap iron with low amounts of phosphorus and sulfur, producing ferrosilicon.[29] Ferrosilicon, an iron-silicon alloy that contains varying ratios of elemental silicon and iron, accounts for about 80% of the world’s production of elemental silicon, with China, the leading supplier of elemental silicon, providing 4.6 million tonnes (or 2/3rds of world output) of silicon, most of it in the form of ferrosilicon. It is followed by Russia (610,000 t), Norway (330,000 t), Brazil (240,000 t), and the United States (170,000 t).[89] Ferrosilicon is primarily used by the iron and steel industry (see below) with primary use as alloying addition in iron or steel and for de-oxidation of steel in integrated steel plants.[29]
Another reaction, sometimes used, is aluminothermal reduction of silicon dioxide, as follows:[90]
- 3 SiO
2 + 4 Al → 3 Si + 2 Al
2O
3
Leaching powdered 96–97% pure silicon with water results in ~98.5% pure silicon, which is used in the chemical industry. However, even greater purity is needed for semiconductor applications, and this is produced from the reduction of tetrachlorosilane (silicon tetrachloride) or trichlorosilane. The former is made by chlorinating scrap silicon and the latter is a byproduct of silicone production. These compounds are volatile and hence can be purified by repeated fractional distillation, followed by reduction to elemental silicon with very pure zinc metal as the reducing agent. The spongy pieces of silicon thus produced are melted and then grown to form cylindrical single crystals, before being purified by zone refining. Other routes use the thermal decomposition of silane or tetraiodosilane (SiI
4). Another process used is the reduction of sodium hexafluorosilicate, a common waste product of the phosphate fertilizer industry, by metallic sodium: this is highly exothermic and hence requires no outside energy source. Hyperfine silicon is made at a higher purity than almost any other material: transistor production requires impurity levels in silicon crystals less than 1 part per 1010, and in special cases impurity levels below 1 part per 1012 are needed and attained.[29]
Silicon nanostructures can directly be produced from silica sand using conventional metalothermic processes, or the combustion synthesis approach. Such nanostructured silicon materials can be used in various functional applications including the anode of lithium ion batteries (LIBs) or phorocatalytic applications.[91]
Applications[edit]
Compounds[edit]
Most silicon is used industrially without being purified, and indeed, often with comparatively little processing from its natural form. More than 90% of the Earth’s crust is composed of silicate minerals, which are compounds of silicon and oxygen, often with metallic ions when negatively charged silicate anions require cations to balance the charge. Many of these have direct commercial uses, such as clays, silica sand, and most kinds of building stone. Thus, the vast majority of uses for silicon are as structural compounds, either as the silicate minerals or silica (crude silicon dioxide). Silicates are used in making Portland cement (made mostly of calcium silicates) which is used in building mortar and modern stucco, but more importantly, combined with silica sand, and gravel (usually containing silicate minerals such as granite), to make the concrete that is the basis of most of the very largest industrial building projects of the modern world.[92]
Silica is used to make fire brick, a type of ceramic. Silicate minerals are also in whiteware ceramics, an important class of products usually containing various types of fired clay minerals (natural aluminium phyllosilicates). An example is porcelain, which is based on the silicate mineral kaolinite. Traditional glass (silica-based soda-lime glass) also functions in many of the same ways, and also is used for windows and containers. In addition, specialty silica based glass fibers are used for optical fiber, as well as to produce fiberglass for structural support and glass wool for thermal insulation.
Silicones often are used in waterproofing treatments, molding compounds, mold-release agents, mechanical seals, high temperature greases and waxes, and caulking compounds. Silicone is also sometimes used in breast implants, contact lenses, explosives and pyrotechnics.[93] Silly Putty was originally made by adding boric acid to silicone oil.[94] Other silicon compounds function as high-technology abrasives and new high-strength ceramics based upon silicon carbide. Silicon is a component of some superalloys.
Alloys[edit]
Elemental silicon is added to molten cast iron as ferrosilicon or silicocalcium alloys to improve performance in casting thin sections and to prevent the formation of cementite where exposed to outside air. The presence of elemental silicon in molten iron acts as a sink for oxygen, so that the steel carbon content, which must be kept within narrow limits for each type of steel, can be more closely controlled. Ferrosilicon production and use is a monitor of the steel industry, and although this form of elemental silicon is grossly impure, it accounts for 80% of the world’s use of free silicon. Silicon is an important constituent of electrical steel, modifying its resistivity and ferromagnetic properties.
The properties of silicon may be used to modify alloys with metals other than iron. «Metallurgical grade» silicon is silicon of 95–99% purity. About 55% of the world consumption of metallurgical purity silicon goes for production of aluminium-silicon alloys (silumin alloys) for aluminium part casts, mainly for use in the automotive industry. Silicon’s importance in aluminium casting is that a significantly high amount (12%) of silicon in aluminium forms a eutectic mixture which solidifies with very little thermal contraction. This greatly reduces tearing and cracks formed from stress as casting alloys cool to solidity. Silicon also significantly improves the hardness and thus wear-resistance of aluminium.[95][96]
Electronics[edit]

Silicon wafer with mirror finish
Most elemental silicon produced remains as a ferrosilicon alloy, and only approximately 20% is refined to metallurgical grade purity (a total of 1.3–1.5 million metric tons/year). An estimated 15% of the world production of metallurgical grade silicon is further refined to semiconductor purity.[96] This typically is the «nine-9» or 99.9999999% purity,[97] nearly defect-free single crystalline material.[98]
Monocrystalline silicon of such purity is usually produced by the Czochralski process, and is used to produce silicon wafers used in the semiconductor industry, in electronics, and in some high-cost and high-efficiency photovoltaic applications.[99] Pure silicon is an intrinsic semiconductor, which means that unlike metals, it conducts electron holes and electrons released from atoms by heat; silicon’s electrical conductivity increases with higher temperatures. Pure silicon has too low a conductivity (i.e., too high a resistivity) to be used as a circuit element in electronics. In practice, pure silicon is doped with small concentrations of certain other elements, which greatly increase its conductivity and adjust its electrical response by controlling the number and charge (positive or negative) of activated carriers. Such control is necessary for transistors, solar cells, semiconductor detectors, and other semiconductor devices used in the computer industry and other technical applications.[100] In silicon photonics, silicon may be used as a continuous wave Raman laser medium to produce coherent light.[101]
In common integrated circuits, a wafer of monocrystalline silicon serves as a mechanical support for the circuits, which are created by doping and insulated from each other by thin layers of silicon oxide, an insulator that is easily produced on Si surfaces by processes of thermal oxidation or local oxidation (LOCOS), which involve exposing the element to oxygen under the proper conditions that can be predicted by the Deal–Grove model. Silicon has become the most popular material for both high power semiconductors and integrated circuits because it can withstand the highest temperatures and greatest electrical activity without suffering avalanche breakdown (an electron avalanche is created when heat produces free electrons and holes, which in turn pass more current, which produces more heat). In addition, the insulating oxide of silicon is not soluble in water, which gives it an advantage over germanium (an element with similar properties which can also be used in semiconductor devices) in certain fabrication techniques.[102]
Monocrystalline silicon is expensive to produce, and is usually justified only in production of integrated circuits, where tiny crystal imperfections can interfere with tiny circuit paths. For other uses, other types of pure silicon may be employed. These include hydrogenated amorphous silicon and upgraded metallurgical-grade silicon (UMG-Si) used in the production of low-cost, large-area electronics in applications such as liquid crystal displays and of large-area, low-cost, thin-film solar cells. Such semiconductor grades of silicon are either slightly less pure or polycrystalline rather than monocrystalline, and are produced in comparable quantities as the monocrystalline silicon: 75,000 to 150,000 metric tons per year. The market for the lesser grade is growing more quickly than for monocrystalline silicon. By 2013, polycrystalline silicon production, used mostly in solar cells, was projected to reach 200,000 metric tons per year, while monocrystalline semiconductor grade silicon was expected to remain less than 50,000 tons per year.[96]
Quantum dots[edit]
Silicon quantum dots are created through the thermal processing of hydrogen silsesquioxane into nanocrystals ranging from a few nanometers to a few microns, displaying size dependent luminescent properties.[103][104] The nanocrystals display large Stokes shifts converting photons in the ultra-violet range to photons in the visible or infrared, depending on the particle size, allowing for applications in quantum dot displays and luminescent solar concentrators due to their limited self absorption. A benefit of using silicon based quantum dots over cadmium or indium is the non-toxic, metal-free nature of silicon.[105][106]
Another application of silicon quantum dots is for sensing of hazardous materials. The sensors take advantage of the luminescent properties of the quantum dots through quenching of the photoluminescence in the presence of the hazardous substance.[107] There are many methods used for hazardous chemical sensing with a few being electron transfer, fluorescence resonance energy transfer, and photocurrent generation.[108] Electron transfer quenching occurs when the lowest unoccupied molecular orbital (LUMO) is slightly lower in energy than the conduction band of the quantum dot, allowing for the transfer electrons between the two, preventing recombination of the holes and electrons within the nanocrystals. The effect can also be achieved in reverse with a donor molecule having its highest occupied molecular orbital (HOMO) slightly higher than a valence band edge of the quantum dot, allowing electrons to transfer between them, filling the holes and preventing recombination. Fluorescence resonance energy transfer occurs when a complex forms between the quantum dot and a quencher molecule. The complex will continue to absorb light but when the energy is converted to the ground state it does not release a photon, quenching the material. The third method uses different approach by measuring the photocurrent emitted by the quantum dots instead of monitoring the photoluminescent display. If the concentration of the desired chemical increases then the photocurrent given off by the nanocrystals will change in response.[109]
Biological role[edit]

A diatom, enclosed in a silica cell wall
Although silicon is readily available in the form of silicates, very few organisms use it directly. Diatoms, radiolaria, and siliceous sponges use biogenic silica as a structural material for their skeletons. Some plants accumulate silica in their tissues and require silicon for their growth, for example rice. Silicon may be taken up by plants as orthosilicic acid (also known as monosilicic acid) and transported through the xylem, where it forms amorphous complexes with components of the cell wall. This has been shown to improve cell wall strength and structural integrity in some plants, thereby reducing insect herbivory and pathogenic infections. In certain plants, silicon may also upregulate the production of volatile organic compounds and phytohormones which play a significant role in plant defense mechanisms.[110][111][112] In more advanced plants, the silica phytoliths (opal phytoliths) are rigid microscopic bodies occurring in the cell.[113][114][111]
Several horticultural crops are known to protect themselves against fungal plant pathogens with silica, to such a degree that fungicide application may fail unless accompanied by sufficient silicon nutrition. Silicaceous plant defense molecules activate some phytoalexins, meaning some of them are signalling substances producing acquired immunity. When deprived, some plants will substitute with increased production of other defensive substances.[111]
Life on Earth is largely composed of carbon, but astrobiology considers that extraterrestrial life may have other hypothetical types of biochemistry. Silicon is considered an alternative to carbon, as it can create complex and stable molecules with four covalent bonds, required for a DNA-analog, and it is available in large quantities.[115]
Marine microbial influences[edit]
Diatoms uses silicon in the biogenic silica (BSIO
2) form,[116] which is taken up by the silicon transport protein (SIT) to be predominantly used in the cell wall structure as frustules.[117] Silicon enters the ocean in a dissolved form such as silicic acid or silicate.[118] Since diatoms are one of the main users of these forms of silicon, they contribute greatly to the concentration of silicon throughout the ocean. Silicon forms a nutrient-like profile in the ocean due to the diatom productivity in shallow depths.[118] Therefore, less concentration of silicon in the upper ocean and more concentrations of silicon in the deep/lower ocean.
Diatom productivity in the upper ocean contribute to the amount of silicon exported to the lower ocean.[119] When diatom cells are lysed in the upper ocean, their nutrients such as iron, zinc, and silicon, are brought to the lower ocean through a process called marine snow. Marine snow involves the downward transfer of particulate organic matter by vertical mixing of dissolved organic matter.[120] It has been suggested that silicon is considered crucial to diatom productivity and as long as there is silicic acid available for diatoms to use, the diatoms can contribute to other important nutrient concentrations in the deep ocean as well.[121]
In coastal zones, diatoms serve as the major phytoplanktonic organisms and greatly contribute to biogenic silica production. In the open ocean, however, diatoms have a reduced role in global annual silica production. Diatoms in North Atlantic and North Pacific subtropical gyres only contribute about 5-7% of global annual marine silica production. The Southern Ocean produces about one-third of global marine biogenic silica.[87] The Southern Ocean is referred to as having a «biogeochemical divide»[122] since only minuscule amounts of silicon are transported out of this region.
Human nutrition[edit]
There is some evidence that silicon is important to human health for their nail, hair, bone, and skin tissues,[123] for example, in studies that demonstrate that premenopausal women with higher dietary silicon intake have higher bone density, and that silicon supplementation can increase bone volume and density in patients with osteoporosis.[124] Silicon is needed for synthesis of elastin and collagen, of which the aorta contains the greatest quantity in the human body,[125] and has been considered an essential element;[126] nevertheless, it is difficult to prove its essentiality, because silicon is very common, and hence, deficiency symptoms are difficult to reproduce.[127][128]
Silicon is currently under consideration for elevation to the status of a «plant beneficial substance by the Association of American Plant Food Control Officials (AAPFCO).»[129][130]
Safety[edit]
People may be exposed to elemental silicon in the workplace by breathing it in, swallowing it, or having contact with the skin or eye. In the latter two cases, silicon poses a slight hazard as an irritant. It is hazardous if inhaled.[131] The Occupational Safety and Health Administration (OSHA) has set the legal limit for silicon exposure in the workplace as 15 mg/m3 total exposure and 5 mg/m3 respiratory exposure over an eight-hour workday. The National Institute for Occupational Safety and Health (NIOSH) has set a recommended exposure limit (REL) of 10 mg/m3 total exposure and 5 mg/m3 respiratory exposure over an eight-hour workday.[132] Inhalation of crystalline silica dust may lead to silicosis, an occupational lung disease marked by inflammation and scarring in the form of nodular lesions in the upper lobes of the lungs.[133]
See also[edit]
- Amorphous silicon
- Black silicon
- Covalent superconductors
- List of countries by silicon production
- List of silicon producers
- Monocrystalline silicon
- Polycrystalline silicon
- Printed silicon electronics
- Silicene
- Silicon nanowire
- Silicon tombac
- Silicon Valley
- Transistor
References[edit]
- ^ «Standard Atomic Weights: Silicon». CIAAW. 2009.
- ^ «New Type of Zero-Valent Tin Compound». Chemistry Europe. 27 August 2016.
- ^ Ram, R. S.; et al. (1998). «Fourier Transform Emission Spectroscopy of the A2D–X2P Transition of SiH and SiD» (PDF). J. Mol. Spectr. 190 (2): 341–352. doi:10.1006/jmsp.1998.7582. PMID 9668026.
- ^ Eranna, Golla (2014). Crystal Growth and Evaluation of Silicon for VLSI and ULSI. CRC Press. p. 7. ISBN 978-1-4822-3281-3.
- ^ Magnetic susceptibility of the elements and inorganic compounds, in Lide, D. R., ed. (2005). CRC Handbook of Chemistry and Physics (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- ^ Weast, Robert (1984). CRC, Handbook of Chemistry and Physics. Boca Raton, Florida: Chemical Rubber Company Publishing. pp. E110. ISBN 0-8493-0464-4.
- ^ a b c d Hopcroft, Matthew A.; Nix, William D.; Kenny, Thomas W. (2010). «What is the Young’s Modulus of Silicon?». Journal of Microelectromechanical Systems. 19 (2): 229. doi:10.1109/JMEMS.2009.2039697.
- ^ Weeks, Mary Elvira (1932). «The discovery of the elements: XII. Other elements isolated with the aid of potassium and sodium: beryllium, boron, silicon, and aluminum». Journal of Chemical Education. 9 (8): 1386–1412. Bibcode:1932JChEd…9.1386W. doi:10.1021/ed009p1386.
- ^ Voronkov, M. G. (2007). «Silicon era». Russian Journal of Applied Chemistry. 80 (12): 2190. doi:10.1134/S1070427207120397.
- ^ Kamal 2022
- ^ Cutter, Elizabeth G. (1978). Plant Anatomy. Part 1 Cells and Tissues (2nd ed.). London: Edward Arnold. ISBN 978-0-7131-2639-6.
- ^ «Silicon». Encyclopedia Britannica. Retrieved 22 August 2019.
- ^ In his table of the elements, Lavoisier listed five «salifiable earths» (i.e., ores that could be made to react with acids to produce salts (salis = salt, in Latin): chaux (calcium oxide), magnésie (magnesia, magnesium oxide), baryte (barium sulfate), alumine (alumina, aluminium oxide), and silice (silica, silicon dioxide). About these «elements», Lavoisier speculates: «We are probably only acquainted as yet with a part of the metallic substances existing in nature, as all those which have a stronger affinity to oxygen than carbon possesses, are incapable, hitherto, of being reduced to a metallic state, and consequently, being only presented to our observation under the form of oxyds, are confounded with earths. It is extremely probable that barytes, which we have just now arranged with earths, is in this situation; for in many experiments it exhibits properties nearly approaching to those of metallic bodies. It is even possible that all the substances we call earths may be only metallic oxyds, irreducible by any hitherto known process.» – from Lavoisier (1799). Elements of Chemistry. Translated by Robert Kerr (4 ed.). Edinburgh, Scotland: William Creec. p. 218. (The original passage appears in: Lavoisier (1789). Traité Élémentaire de Chimie. Vol. 1. Paris, France: Cuchet. p. 174.
- ^ a b c d e Greenwood & Earnshaw 1997, p. 328.
- ^ Davy, Humphry (1808). «Electro chemical researches, on the decomposition of the earths; with observations on the metals obtained from the alkaline earths, and on the amalgam procured from ammonia». Philosophical Transactions of the Royal Society of London. W. Bowyer and J. Nichols. 98: 333–370.. On p. 353 Davy coins the name «silicium» : «Had I been so fortunate as to have obtained more certain evidences on this subject, and to have procured the metallic substances I was in search of, I should have proposed for them the names of silicium [silicon], alumium [aluminium], zirconium, and glucium [beryllium].»
- ^ «14 Silicon». Elements.vanderkrogt.net. Retrieved 2008-09-12.
- ^ Gay-Lussac, Joseph Louis; Thénard, Louis Jacques baron (1811). Recherches physico-chimiques, faites sur la pile: sur la préparation chimique et les propriétés du potassium et du sodium; sur la décomposition de l’acide boracique; sur les acides fluorique, muriatique et muriatique oxigéné; sur l’action chimique de la lumière; sur l’analyse végétale et animale, etc (in French). Deterville. pp. 313–314; vol. 2, pp. 55–65.
- ^ Thomson, Thomas; Baldwin, Charles; Blackwood, William; Baldwin, Cradock; Bell & Bradfute, bookseller; Hodges & McArthur, bookseller (1817). A system of chemistry : in four volumes. University of Wisconsin — Madison. London : Printed for Baldwin, Craddock, and Joy, Paternoster-Row; William Blackwood, and Bell and Bradfute, Edinburgh; and Hodges and Macarthur, Dublin. p. 252.: «The base of silica has been usually considered as a metal, and called silicium. But as there is not the smallest evidence for its metallic nature, and as it bears a close resemblance to boron and carbon, it is better to class it along with these bodies, and to give it the name of silicon.»
- ^ See
- Berzelius announced his discovery of silicon («silicium») in: Berzelius, J. (presented: 1823; published: 1824) «Undersökning af flusspatssyran och dess märkvärdigaste föreningar» (Investigation of hydrofluoric acid and of its most noteworthy compounds), Kongliga Vetenskaps-Academiens Handlingar [Proceedings of the Royal Science Academy], 12 : 46–98. The isolation of silicon and its characterization are detailed in the section titled «Flussspatssyrad kisseljords sönderdelning med kalium,» pp. 46–68.
- The above article was printed in German in: J.J. Berzelius (1824) «II. Untersuchungen über Flussspathsäure und deren merkwürdigsten Verbindungen» (II. Investigations of hydrofluoric acid and its most noteworthy compounds), Annalen der Physik, 77: 169–230. The isolation of silicon is detailed in the section titled: «Zersetzung der flussspaths. Kieselerde durch Kalium» (Decomposition of silicate fluoride by potassium), pp. 204–210.
- The above article was reprinted in French in: Berzelius (1824) «Décomposition du fluate de silice par le potassium» (Decomposition of silica fluoride by potassium), Annales de Chimie et de Physique, 27: 337–359.
- Reprinted in English in: «On the mode of obtaining silicium, and on the characters and properties of that substance». The Philosophical Magazine and Journal: Comprehending Various Branches of Science, the Liberal and Fine Arts, Agriculture, Manufactures, and Commerce. Richard Taylor and Company. 65: 254–267. 1825.
- ^ Weeks, Mary Elvira (1932). «The discovery of the elements: XII. Other elements isolated with the aid of potassium and sodium: beryllium, boron, silicon, and aluminum». Journal of Chemical Education. 9 (8): 1386–1412. Bibcode:1932JChEd…9.1386W. doi:10.1021/ed009p1386.
- ^ Voronkov, M.G. (2007). «Silicon era». Russian Journal of Applied Chemistry. 80 (12): 2190. doi:10.1134/S1070427207120397. S2CID 195240638.
- ^ a b Kipping, Frederic Stanley (1937-03-01). «The bakerian lecture organic derivatives of silicon». Proceedings of the Royal Society of London. Series A — Mathematical and Physical Sciences. 159 (896): 139–148. Bibcode:1937RSPSA.159..139K. doi:10.1098/rspa.1937.0063.
- ^ Muller, Richard (January 1965). «One hundred years of organosilicon chemistry». Journal of Chemical Education. 42 (1): 41. Bibcode:1965JChEd..42…41M. doi:10.1021/ed042p41. ISSN 0021-9584.
- ^ In 1854, Deville was trying to prepare aluminium metal from aluminium chloride that was heavily contaminated with silicon chloride. Deville used two methods to prepare aluminium: heating aluminium chloride with sodium metal in an inert atmosphere (of hydrogen); and melting aluminum chloride with sodium chloride and then electrolyzing the mixture. In both cases, pure silicon was produced: the silicon dissolved in the molten aluminium, but crystallized upon cooling. Dissolving the crude aluminum in hydrochloric acid revealed flakes of crystallized silicon. See: Henri Sainte-Claire Deville (1854) «Note sur deux procédés de préparation de l’aluminium et sur une nouvelle forme du silicium» (Note on two procedures for the preparation of aluminium and on a new form of silicon), Comptes rendus, 39: 321–326.
Subsequently, Deville obtained crystalline silicon by heating the chloride or fluoride of silicon with sodium metal, isolating the amorphous silicon, then melting the amorphous form with salt and heating the mixture until most of the salt evaporated. See: Sainte-Claire Deville, H. (1855). «Du silicium et du titane (On silicon and titanium)». Comptes rendus. 40: 1034–1036. - ^ «Information on silicon – history, thermodynamic, chemical, physical and electronic properties». Etacude. Retrieved 2021-06-08.
- ^ «Silicon: History». Nautilus.fis.uc.pt. 2011-07-27. Archived from the original on July 27, 2011.
- ^ a b Aufray, B.; Kara, A.; Vizzini, S. B.; Oughaddou, H.; LéAndri, C.; Ealet, B.; Le Lay, G. (2010). «Graphene-like silicon nanoribbons on Ag(110): A possible formation of silicene». Applied Physics Letters. 96 (18): 183102. Bibcode:2010ApPhL..96r3102A. doi:10.1063/1.3419932.
- ^ a b Lalmi, B.; Oughaddou, H.; Enriquez, H.; Kara, A.; Vizzini, S. B.; Ealet, B. N.; Aufray, B. (2010). «Epitaxial growth of a silicene sheet». Applied Physics Letters. 97 (22): 223109. arXiv:1204.0523. Bibcode:2010ApPhL..97v3109L. doi:10.1063/1.3524215. S2CID 118490651.
- ^ a b c d e f g h i j k l m n Greenwood & Earnshaw 1997, p. 330.
- ^ Greenwood & Earnshaw 1997, pp. 337–340
- ^ a b «Timeline». The Silicon Engine. Computer History Museum. Retrieved 22 August 2019.
- ^ a b «1901: Semiconductor Rectifiers Patented as «Cat’s Whisker» Detectors». The Silicon Engine. Computer History Museum. Retrieved 23 August 2019.
- ^ «1947: Invention of the Point-Contact Transistor». The Silicon Engine. Computer History Museum. Retrieved 23 August 2019.
- ^ «1954: Morris Tanenbaum fabricates the first silicon transistor at Bell Labs». The Silicon Engine. Computer History Museum. Retrieved 23 August 2019.
- ^ Bassett, Ross Knox (2007). To the Digital Age: Research Labs, Start-up Companies, and the Rise of MOS Technology. Johns Hopkins University Press. pp. 22–23. ISBN 978-0-8018-8639-3.
- ^ Saxena, A. (2009). Invention of integrated circuits: untold important facts. International series on advances in solid state electronics and technology. World Scientific. pp. 96–97. ISBN 978-981-281-445-6.
- ^ a b Feldman, Leonard C. (2001). «Introduction». Fundamental Aspects of Silicon Oxidation. Springer Science & Business Media. pp. 1–11. ISBN 978-3-540-41682-1.
- ^ Dabrowski, Jarek; Müssig, Hans-Joachim (2000). «1.2. The Silicon Age». Silicon Surfaces and Formation of Interfaces: Basic Science in the Industrial World. World Scientific. pp. 3–13. ISBN 978-981-02-3286-3.
- ^ Siffert, Paul; Krimmel, Eberhard (2013). «Preface». Silicon: Evolution and Future of a Technology. Springer Science & Business Media. ISBN 978-3-662-09897-4.
- ^ Uskali, T.; Nordfors, D. (23 May 2007). «The role of journalism in creating the metaphor of Silicon Valley» (PDF). Innovation Journalism 4 Conference, Stanford University. Archived from the original (PDF) on 2012-09-07. Retrieved 2016-08-08.
- ^ «Silicon and Germanium». hyperphysics.phy-astr.gsu.edu. Retrieved 2021-06-07.
- ^ King 1995, pp. xiii–xviii
- ^ Greenwood & Earnshaw 1997, pp. 372.
- ^ a b c d e f g h i j Greenwood & Earnshaw 1997, p. 331.
- ^ Vladimir E. Dmitrienko and Viacheslav A. Chizhikov (2020). «An infinite family of bc8-like metastable phases in silicon». Phys. Rev. B. 101 (24): 245203. arXiv:1912.10672. Bibcode:2020PhRvB.101x5203D. doi:10.1103/PhysRevB.101.245203. S2CID 209444444.
- ^ a b c d e f g Audi, G.; Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S. (2017). «The NUBASE2016 evaluation of nuclear properties» (PDF). Chinese Physics C. 41 (3): 030001. Bibcode:2017ChPhC..41c0001A. doi:10.1088/1674-1137/41/3/030001.
- ^ Jerschow, Alexej. «Interactive NMR Frequency Map». New York University. Retrieved 2011-10-20.
- ^ Seitenzahl, Ivo Rolf; Townsley, Dean M. (2017). Nucleosynthesis in Thermonuclear Supernovae. Handbook of Supernovae. pp. 1955–1978. arXiv:1704.00415. Bibcode:2017hsn..book.1955S. doi:10.1007/978-3-319-21846-5_87. ISBN 978-3-319-21845-8. S2CID 118993185.
- ^ Khokhlov, A. M.; Oran, E. S.; Wheeler, J. C. (April 1997). «Deflagration‐to‐Detonation Transition in Thermonuclear Supernovae». The Astrophysical Journal. 478 (2): 678–688. arXiv:astro-ph/9612226. Bibcode:1997ApJ…478..678K. doi:10.1086/303815. S2CID 53486905.
- ^ Cameron, A.G.W. (1973). «Abundance of the Elements in the Solar System» (PDF). Space Science Reviews. 15 (1): 121–146. Bibcode:1973SSRv…15..121C. doi:10.1007/BF00172440. S2CID 120201972. Archived from the original (PDF) on 2011-10-21.
- ^ Reynolds, B. C. (June 2009). «Modeling the modern marine δ 30 Si distribution: MODELING THE MODERN MARINE δ 30 Si DISTRIBUTION». Global Biogeochemical Cycles. 23 (2): 1–13. doi:10.1029/2008GB003266. S2CID 128652214.
- ^ Stapf, André; Gondek, Christoph; Kroke, Edwin; Roewer, Gerhard (2019), Yang, Deren (ed.), «Wafer Cleaning, Etching, and Texturization», Handbook of Photovoltaic Silicon, Berlin, Heidelberg: Springer Berlin Heidelberg, pp. 311–358, doi:10.1007/978-3-662-56472-1_17, ISBN 978-3-662-56471-4, S2CID 226945433, retrieved 2021-03-07
- ^ a b Grabmaier, J. (1982). Silicon Chemical Etching. Berlin, Heidelberg: Springer Berlin Heidelberg. ISBN 978-3-642-68765-5. OCLC 840294227.
- ^ Greenwood & Earnshaw 1997, pp. 331–5
- ^ Kaupp, Martin (1 December 2006). «The role of radial nodes of atomic orbitals for chemical bonding and the periodic table» (PDF). Journal of Computational Chemistry. 28 (1): 320–325. doi:10.1002/jcc.20522. PMID 17143872. S2CID 12677737. Retrieved 14 October 2016.
- ^ a b c King 1995, pp. 43–44
- ^ a b Greenwood & Earnshaw 1997, pp. 374.
- ^ Kaupp, Martin (2007). «The role of radial nodes of atomic orbitals for chemical bonding and the periodic table». Journal of Computational Chemistry. 28 (1): 320–325. doi:10.1002/jcc.20522. ISSN 0192-8651. PMID 17143872.
- ^ Greenwood & Earnshaw 1997, pp. 327–328.
- ^ a b Greenwood & Earnshaw 1997, pp. 359–361.
- ^ Greenwood & Earnshaw 1997, p. 335-337.
- ^ King 1995, pp. 45–47
- ^ Wiber, E. (1977). «Alfred Stock and the Renaissance of Inorganic Chemistry» (PDF). Pure Appl. Chem. 49 (6): 691–700. doi:10.1351/pac197749060691. S2CID 53313463.
- ^ Mellor, J.W. (1947). A Comprehensive Treatise on Inorganic and Theoretical Chemistry. Vol. VI, [C(Part II), Si, Silicates]. Longman, Green and Co. pp. 223–7. OCLC 1044702591.
- ^ Porterfield, W.W. (2013) [1993]. «4.8 Bonding in Elements». Inorganic Chemistry: A Unified Approach (2nd ed.). Elsevier. p. 219. ISBN 978-0-323-13894-9.
- ^ Wiberg, N.; Wiberg, E.; Holleman, A.F. (2001). «2.2.3 Higher Saturated Silanes». Inorganic Chemistry. Academic Press. p. 844. ISBN 0-12-352651-5.
- ^ King 1995, p. 47
- ^ Miller, R.D.; Michl, J. (1989). «Polysilane high polymers». Chemical Reviews. 89 (6): 1359. doi:10.1021/cr00096a006.
- ^ a b c d Greenwood & Earnshaw 1997, pp. 340.
- ^ a b c King 1995, p. 48
- ^ Greenwood & Earnshaw 1997, p. 342-347.
- ^ a b c Greenwood & Earnshaw 1997, p. 342.
- ^ a b c d e Greenwood & Earnshaw 1997, p. 347.
- ^ Geological Survey (U.S.) (1975). Geological Survey professional paper.
- ^ Korzhinsky, M.A.; Tkachenko, S.I.; Shmulovich, K.I.; Steinberg, G.S. (1995). «Native AI and Si formation» (PDF). Nature. 375 (6532): 544. Bibcode:1995Natur.375..544K. doi:10.1038/375544a0. ISSN 0028-0836. S2CID 39954119.
- ^ Cordua, Courtesy of Dr Bill (1998-01-10), English: PDF file entitled: «Silicon, Silica, Silicates and Silicone» (PDF), archived from the original (PDF) on 2016-04-18, retrieved 2016-03-29
- ^ Greenwood & Earnshaw 1997, p. 347-359.
- ^ a b c d Greenwood & Earnshaw 1997, p. 359.
- ^ a b c Greenwood & Earnshaw 1997, p. 334.
- ^ Cheung, Rebecca (2006). Silicon carbide microelectromechanical systems for harsh environments. Imperial College Press. p. 3. ISBN 978-1-86094-624-0.
- ^ Morkoç, H.; Strite, S.; Gao, G.B.; Lin, M.E.; Sverdlov, B.; Burns, M. (1994). «Large-band-gap SiC, III–V nitride, and II–VI ZnSe-based semiconductor device technologies». Journal of Applied Physics. 76 (3): 1363. Bibcode:1994JAP….76.1363M. doi:10.1063/1.358463.
- ^ a b c d e Greenwood & Earnshaw 1997, p. 361.
- ^ a b Clayden, pp. 668–77
- ^ Greenwood & Earnshaw 1997, p. 366.
- ^ a b Greenwood & Earnshaw 1997, p. 329.
- ^ Greenwood & Earnshaw 1997, pp. 329–330
- ^ a b c Tréguer, Paul J.; De La Rocha, Christina L. (3 January 2013). «The World Ocean Silica Cycle». Annual Review of Marine Science. 5 (1): 477–501. doi:10.1146/annurev-marine-121211-172346. PMID 22809182.
- ^ a b Tegen, Ina; Kohfeld, Karen (2006). Atmospheric transport of silicon. Island Press. pp. 81–91. ISBN 1-59726-115-7.
- ^ «Silicon Commodities Report 2011» (PDF). USGS. Retrieved 2011-10-20.
- ^ Zulehner, Neuer & Rau, p. 574
- ^ Kamali, A.R. (2019). «Ultra-fast shock-wave combustion synthesis of nanostructured silicon from sand with excellent Li storage performance». Sustainable Energy & Fuels. 3 (6): 1396–1405. doi:10.1039/C9SE00046A. S2CID 139986478.
- ^ Greenwood & Earnshaw 1997, p. 356.
- ^ Koch, E.C.; Clement, D. (2007). «Special Materials in Pyrotechnics: VI. Silicon – An Old Fuel with New Perspectives». Propellants, Explosives, Pyrotechnics. 32 (3): 205. doi:10.1002/prep.200700021.
- ^ Walsh, Tim (2005). «Silly Putty». Timeless toys: classic toys and the playmakers who created them. Andrews McMeel Publishing. ISBN 978-0-7407-5571-2.
- ^ Apelian, D. (2009). «Aluminum Cast Alloys: Enabling Tools for Improved Performance» (PDF). Wheeling, Illinois: North American Die Casting Association. Archived from the original (PDF) on 2012-01-06.
- ^ a b c Corathers, Lisa A. 2009 Minerals Yearbook. USGS
- ^ «Semi» SemiSource 2006: A supplement to Semiconductor International. December 2005. Reference Section: How to Make a Chip. Adapted from Design News. Reed Electronics Group.
- ^ SemiSource 2006: A supplement to Semiconductor International. December 2005. Reference Section: How to Make a Chip. Adapted from Design News. Reed Electronics Group.
- ^ Zulehner, Neuer & Rau, p. 590
- ^ Zulehner, Neuer & Rau, p. 573
- ^ Dekker, R; Usechak, N; Först, M; Driessen, A (2008). «Ultrafast nonlinear all-optical processes in silicon-on-insulator waveguides». Journal of Physics D. 40 (14): R249–R271. Bibcode:2007JPhD…40..249D. doi:10.1088/0022-3727/40/14/r01. S2CID 123008652.
- ^ Semiconductors Without the Quantum Physics. Electropaedia
- ^ Clark, Rhett J.; Aghajamali, Maryam; Gonzalez, Christina M.; Hadidi, Lida; Islam, Muhammad Amirul; Javadi, Morteza; Mobarok, Md Hosnay; Purkait, Tapas K.; Robidillo, Christopher Jay T.; Sinelnikov, Regina; Thiessen, Alyxandra N. (2017-01-10). «From Hydrogen Silsesquioxane to Functionalized Silicon Nanocrystals». Chemistry of Materials. 29 (1): 80–89. doi:10.1021/acs.chemmater.6b02667. ISSN 0897-4756.
- ^ Hessel, Colin M.; Henderson, Eric J.; Veinot, Jonathan G. C. (2007). «Hydrogen Silsesquioxane: A Molecular Precursor for Nanocrystalline Si—SiO2 Composites and Freestanding Hydride-Surface-Terminated Silicon Nanoparticles». ChemInform. 38 (10). doi:10.1002/chin.200710014. ISSN 1522-2667.
- ^ Lim, Cheol Hong; Han, Jeong-Hee; Cho, Hae-Won; Kang, Mingu (2014). «Studies on the Toxicity and Distribution of Indium Compounds According to Particle Size in Sprague-Dawley Rats». Toxicological Research. 30 (1): 55–63. doi:10.5487/TR.2014.30.1.055. ISSN 1976-8257. PMC 4007045. PMID 24795801.
- ^ Zou, Hui; Wang, Tao; Yuan, Junzhao; Sun, Jian; Yuan, Yan; Gu, Jianhong; Liu, Xuezhong; Bian, Jianchun; Liu, Zongping (2020-03-15). «Cadmium-induced cytotoxicity in mouse liver cells is associated with the disruption of autophagic flux via inhibiting the fusion of autophagosomes and lysosomes». Toxicology Letters. 321: 32–43. doi:10.1016/j.toxlet.2019.12.019. ISSN 0378-4274. PMID 31862506. S2CID 209435190.
- ^ Nguyen, An; Gonzalez, Christina M; Sinelnikov, Regina; Newman, W; Sun, Sarah; Lockwood, Ross; Veinot, Jonathan G C; Meldrum, Al (2016-02-10). «Detection of nitroaromatics in the solid, solution, and vapor phases using silicon quantum dot sensors». Nanotechnology. 27 (10): 105501. Bibcode:2016Nanot..27j5501N. doi:10.1088/0957-4484/27/10/105501. ISSN 0957-4484. PMID 26863492. S2CID 24292648.
- ^ Gonzalez, Christina M.; Veinot, Jonathan G. C. (2016-06-02). «Silicon nanocrystals for the development of sensing platforms». Journal of Materials Chemistry C. 4 (22): 4836–4846. doi:10.1039/C6TC01159D. ISSN 2050-7534.
- ^ Yue, Zhao; Lisdat, Fred; Parak, Wolfgang J.; Hickey, Stephen G.; Tu, Liping; Sabir, Nadeem; Dorfs, Dirk; Bigall, Nadja C. (2013-04-24). «Quantum-Dot-Based Photoelectrochemical Sensors for Chemical and Biological Detection». ACS Applied Materials & Interfaces. 5 (8): 2800–2814. doi:10.1021/am3028662. ISSN 1944-8244. PMID 23547912.
- ^ Kim, Sang Gyu; Kim, Ki Woo; Park, Eun Woo; Choi, Doil (2002). «Silicon-Induced Cell Wall Fortification of Rice Leaves: A Possible Cellular Mechanism of Enhanced Host Resistance to Blast». Phytopathology. 92 (10): 1095–103. doi:10.1094/PHYTO.2002.92.10.1095. PMID 18944220.
- ^ a b c Epstein, Emanuel (1999). «SILICON». Annual Review of Plant Physiology and Plant Molecular Biology. 50: 641–664. doi:10.1146/annurev.arplant.50.1.641. PMID 15012222.
- ^ Leroy, Nicolas; de Tombeur, Felix; Walgraffe, Yseult; Cornelis, Jean-Thomas; Verheggen, Francois (23 October 2019). «Silicon and plant natural defenses against insect pests: impact on plant volatile organic compounds and cascade effects on multitrophic interactions». Plants. 8 (444): 444. doi:10.3390/plants8110444. PMC 6918431. PMID 31652861.
- ^ Rahman, Atta-ur- (2008). «Silicon». Studies in Natural Products Chemistry. Vol. 35. p. 856. ISBN 978-0-444-53181-0.
- ^ Exley, C. (1998). «Silicon in life:A bioinorganic solution to bioorganic essentiality». Journal of Inorganic Biochemistry. 69 (3): 139–144. doi:10.1016/S0162-0134(97)10010-1.
- ^ Aguilera Mochón, Juan Antonio (2016). La vida no terrestre [The non-terrestrial life] (in Spanish). RBA. pp. 43–45. ISBN 978-84-473-8665-9.
- ^ Bidle, Kay D.; Manganelli, Maura; Azam, Farooq (2002-12-06). «Regulation of Oceanic Silicon and Carbon Preservation by Temperature Control on Bacteria». Science. 298 (5600): 1980–1984. Bibcode:2002Sci…298.1980B. doi:10.1126/science.1076076. ISSN 0036-8075. PMID 12471255. S2CID 216994.
- ^ Durkin, Colleen A.; Koester, Julie A.; Bender, Sara J.; Armbrust, E. Virginia (2016). «The evolution of silicon transporters in diatoms». Journal of Phycology. 52 (5): 716–731. doi:10.1111/jpy.12441. ISSN 1529-8817. PMC 5129515. PMID 27335204.
- ^ a b Dugdale, R. C.; Wilkerson, F. P. (2001-12-30). «Sources and fates of silicon in the ocean: the role of diatoms in the climate and glacial cycles». Scientia Marina. 65 (S2): 141–152. doi:10.3989/scimar.2001.65s2141. ISSN 1886-8134.
- ^ Baines, Stephen B.; Twining, Benjamin S.; Brzezinski, Mark A.; Krause, Jeffrey W.; Vogt, Stefan; Assael, Dylan; McDaniel, Hannah (December 2012). «Significant silicon accumulation by marine picocyanobacteria». Nature Geoscience. 5 (12): 886–891. Bibcode:2012NatGe…5..886B. doi:10.1038/ngeo1641. ISSN 1752-0908.
- ^ Turner, Jefferson T. (January 2015). «Zooplankton fecal pellets, marine snow, phytodetritus and the ocean’s biological pump». Progress in Oceanography. 130: 205–248. Bibcode:2015PrOce.130..205T. doi:10.1016/j.pocean.2014.08.005. ISSN 0079-6611.
- ^ Yool, Andrew; Tyrrell, Toby (2003). «Role of diatoms in regulating the ocean’s silicon cycle». Global Biogeochemical Cycles. 17 (4): n/a. Bibcode:2003GBioC..17.1103Y. CiteSeerX 10.1.1.394.3912. doi:10.1029/2002GB002018. ISSN 1944-9224. S2CID 16849373.
- ^ Marinov, I.; Gnanadesikan, A.; Toggweiler, J. R.; Sarmiento, J. L. (June 2006). «The Southern Ocean biogeochemical divide». Nature. 441 (7096): 964–967. Bibcode:2006Natur.441..964M. doi:10.1038/nature04883. PMID 16791191. S2CID 4428683.
- ^ Martin, Keith R. (2013). «Chapter 14. Silicon: The Health Benefits of a Metalloid». In Astrid Sigel; Helmut Sigel; Roland K.O. Sigel (eds.). Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. Vol. 13. Springer. pp. 451–473. doi:10.1007/978-94-007-7500-8_14. ISBN 978-94-007-7499-5. PMID 24470100.
- ^ Jugdaohsingh, R. (Mar–Apr 2007). «Silicon and bone health». The Journal of Nutrition, Health and Aging. 11 (2): 99–110. PMC 2658806. PMID 17435952.
- ^ Loeper, J.; Fragny, M. (1978). The Physiological Role of the Silicon and its AntiAtheromatous Action. Biochemistry of Silicon and Related Problems. pp. 281–296. doi:10.1007/978-1-4613-4018-8_13. ISBN 978-1-4613-4020-1.
- ^ Nielsen, Forrest H. (1984). «Ultratrace Elements in Nutrition». Annual Review of Nutrition. 4: 21–41. doi:10.1146/annurev.nu.04.070184.000321. PMID 6087860.
- ^ Lippard, Stephen J.; Jeremy M. Berg (1994). Principles of Bioinorganic Chemistry. Mill Valley, CA: University Science Books. p. 411. ISBN 978-0-935702-72-9.
- ^ Muhammad Ansar Farooq; Karl-Josef Dietz (2015). «Silicon as Versatile Player in Plant and Human Biology: Overlooked and Poorly Understood Muhammad Ansar Farooq and Karl-J». Front. Plant Sci. 6 (994): 994. doi:10.3389/fpls.2015.00994. PMC 4641902. PMID 26617630.
- ^ «AAPFCO Board of Directors 2006 Mid-Year Meeting» (PDF). Association of American Plant Food Control Officials. Archived from the original (PDF) on 6 January 2012. Retrieved 2011-07-18.
A presentation was made for Excell Minerals to recognize Silicon as a recognized plant nutrient
- ^ Miranda, Stephen R.; Barker, Bruce (August 4, 2009). «Silicon: Summary of Extraction Methods». Harsco Minerals. Archived from the original on November 12, 2012. Retrieved 2011-07-18.
- ^ Science Lab.com. «Material Safety Data Sheet: Silicon MSDS». sciencelab.com. Archived from the original on 23 March 2018. Retrieved 11 March 2018.
- ^ «CDC – NIOSH Pocket Guide to Chemical Hazards – Silicon». www.cdc.gov. Retrieved 2015-11-21.
- ^ Jane A. Plant; Nick Voulvoulis; K. Vala Ragnarsdottir (2012). Pollutants, Human Health and the Environment: A Risk Based Approach. Applied Geochemistry. Vol. 26. John Wiley & Sons. p. 273. Bibcode:2011ApGC…26S.238P. doi:10.1016/j.apgeochem.2011.03.113. ISBN 978-0-470-74261-7. Retrieved 24 August 2012.
Bibliography[edit]
- Clayden, Jonathan; Greeves, Nick; Warren, Stuart (2012). Organic Chemistry (2nd ed.). Oxford University Press. ISBN 978-0-19-927029-3.
- Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 978-0-08-037941-8.
- King, R. Bruce (1995). Inorganic Chemistry of Main Group Elements. Wiley-VCH. ISBN 978-0-471-18602-1.
- Zulehner, Werner; Neuer, Bernd; Rau, Gerhard. «Silicon». Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a23_721.
- Kamal, Kamal Y. (2022). «The Silicon Age: Trends in Semiconductor Devices Industry» (PDF). Journal of Engineering Science and Technology Review. 15 (1): 110–5. doi:10.25103/jestr.151.14. S2CID 249074588.
External links[edit]
- «Silicon Video — The Periodic Table of Videos — University of Nottingham». www.periodicvideos.com. Retrieved 2021-06-08.
- «CDC — NIOSH Pocket Guide to Chemical Hazards — Silicon». www.cdc.gov. Retrieved 2021-06-08.
- «Physical properties of Silicon (Si)». www.ioffe.ru. Retrieved 2021-06-08.
- The Story of Solar-Grade Silicon. Asianometry. 30 November 2022.
Not to be confused with the silicon-containing synthetic polymer silicone.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Silicon | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Allotropes | see Allotropes of silicon | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Appearance | crystalline, reflective with bluish-tinged faces | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard atomic weight Ar°(Si) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Silicon in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | group 14 (carbon group) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block | p-block | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Ne] 3s2 3p2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | 1687 K (1414 °C, 2577 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boiling point | 3538 K (3265 °C, 5909 °F) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density (near r.t.) | 2.3290 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| when liquid (at m.p.) | 2.57 g/cm3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | 50.21 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of vaporization | 383 kJ/mol | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | 19.789 J/(mol·K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapor pressure
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | −4, −3, −2, −1, 0,[2] +1,[3] +2, +3, +4 (an amphoteric oxide) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.90 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionization energies |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic radius | empirical: 111 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | 111 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Van der Waals radius | 210 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Spectral lines of silicon |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | primordial | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | face-centered diamond-cubic
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Speed of sound thin rod | 8433 m/s (at 20 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal expansion | 2.6 µm/(m⋅K) (at 25 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | 149 W/(m⋅K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrical resistivity | 2.3×103 Ω⋅m (at 20 °C)[4] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Band gap | 1.12 eV (at 300 K) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | diamagnetic[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar magnetic susceptibility | −3.9×10−6 cm3/mol (298 K)[6] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Young’s modulus | 130–188 GPa[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Shear modulus | 51–80 GPa[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bulk modulus | 97.6 GPa[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Poisson ratio | 0.064–0.28[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mohs hardness | 6.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 7440-21-3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Naming | after Latin silex or silicis, meaning ‘flint’ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Prediction | Antoine Lavoisier (1787) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery and first isolation | Jöns Jacob Berzelius[8][9] (1823) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Named by | Thomas Thomson (1817) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Isotopes of silicon
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| references |
Silicon is a chemical element with the symbol Si and atomic number 14. It is a hard, brittle crystalline solid with a blue-grey metallic luster, and is a tetravalent metalloid and semiconductor. It is a member of group 14 in the periodic table: carbon is above it; and germanium, tin, lead, and flerovium are below it. It is relatively unreactive.
Because of its high chemical affinity for oxygen, it was not until 1823 that Jöns Jakob Berzelius was first able to prepare it and characterize it in pure form. Its oxides form a family of anions known as silicates. Its melting and boiling points of 1414 °C and 3265 °C, respectively, are the second highest among all the metalloids and nonmetals, being surpassed only by boron.
Silicon is the eighth most common element in the universe by mass, but very rarely occurs as the pure element in the Earth’s crust. It is widely distributed in space in cosmic dusts, planetoids, and planets as various forms of silicon dioxide (silica) or silicates. More than 90% of the Earth’s crust is composed of silicate minerals, making silicon the second most abundant element in the Earth’s crust (about 28% by mass), after oxygen.
Most silicon is used commercially without being separated, often with very little processing of the natural minerals. Such use includes industrial construction with clays, silica sand, and stone. Silicates are used in Portland cement for mortar and stucco, and mixed with silica sand and gravel to make concrete for walkways, foundations, and roads. They are also used in whiteware ceramics such as porcelain, and in traditional silicate-based soda-lime glass and many other specialty glasses. Silicon compounds such as silicon carbide are used as abrasives and components of high-strength ceramics. Silicon is the basis of the widely used synthetic polymers called silicones.
The late 20th century to early 21st century has been described as the Silicon Age (also known as the Digital Age or Information Age) because of the large impact that elemental silicon has on the modern world economy. The small portion of very highly purified elemental silicon used in semiconductor electronics (<10%[citation needed]) is essential to the transistors and integrated circuit chips used in most modern technology such as smartphones and other computers. In 2019, 32.4% of the semiconductor market segment was for networks and communications devices, and the semiconductors industry is projected to reach $726.73 billion by 2027. [10]
Silicon is an essential element in biology. Only traces are required by most animals, but some sea sponges and microorganisms, such as diatoms and radiolaria, secrete skeletal structures made of silica. Silica is deposited in many plant tissues.[11]
History[edit]
Owing to the abundance of silicon in the Earth’s crust, natural silicon-based materials have been used for thousands of years. Silicon rock crystals were familiar to various ancient civilizations, such as the predynastic Egyptians who used it for beads and small vases, as well as the ancient Chinese. Glass containing silica was manufactured by the Egyptians since at least 1500 BC, as well as by the ancient Phoenicians. Natural silicate compounds were also used in various types of mortar for construction of early human dwellings.[12]
Discovery[edit]
In 1787, Antoine Lavoisier suspected that silica might be an oxide of a fundamental chemical element,[13] but the chemical affinity of silicon for oxygen is high enough that he had no means to reduce the oxide and isolate the element.[14] After an attempt to isolate silicon in 1808, Sir Humphry Davy proposed the name «silicium» for silicon, from the Latin silex, silicis for flint, and adding the «-ium» ending because he believed it to be a metal.[15] Most other languages use transliterated forms of Davy’s name, sometimes adapted to local phonology (e.g. German Silizium, Turkish silisyum, Catalan silici, Armenian Սիլիցիում or Silitzioum). A few others use instead a calque of the Latin root (e.g. Russian кремний, from кремень «flint»; Greek πυρίτιο from πυρ «fire»; Finnish pii from piikivi «flint», Czech křemík from křemen «quartz», «flint»).[16]
Gay-Lussac and Thénard are thought to have prepared impure amorphous silicon in 1811, through the heating of recently isolated potassium metal with silicon tetrafluoride, but they did not purify and characterize the product, nor identify it as a new element.[17] Silicon was given its present name in 1817 by Scottish chemist Thomas Thomson. He retained part of Davy’s name but added «-on» because he believed that silicon was a nonmetal similar to boron and carbon.[18] In 1824, Jöns Jacob Berzelius prepared amorphous silicon using approximately the same method as Gay-Lussac (reducing potassium fluorosilicate with molten potassium metal), but purifying the product to a brown powder by repeatedly washing it.[19] As a result, he is usually given credit for the element’s discovery.[20][21] The same year, Berzelius became the first to prepare silicon tetrachloride; silicon tetrafluoride had already been prepared long before in 1771 by Carl Wilhelm Scheele by dissolving silica in hydrofluoric acid.[14] In 1823 for the first time Jacob Berzelius discovered silicon tetrachloride (SiCl4).[22] In 1846 Von Ebelman’s had synthesized Tetraethyl orthosilicate (Si(OC2H5)4).[23][22]
Silicon in its more common crystalline form was not prepared until 31 years later, by Deville.[24][25] By electrolyzing a mixture of sodium chloride and aluminium chloride containing approximately 10% silicon, he was able to obtain a slightly impure allotrope of silicon in 1854.[26] Later, more cost-effective methods have been developed to isolate several allotrope forms, the most recent being silicene in 2010.[27][28] Meanwhile, research on the chemistry of silicon continued; Friedrich Wöhler discovered the first volatile hydrides of silicon, synthesising trichlorosilane in 1857 and silane itself in 1858, but a detailed investigation of the silanes was only carried out in the early 20th century by Alfred Stock, despite early speculation on the matter dating as far back as the beginnings of synthetic organic chemistry in the 1830s.[29][30] Similarly, the first organosilicon compound, tetraethylsilane, was synthesised by Charles Friedel and James Crafts in 1863, but detailed characterisation of organosilicon chemistry was only done in the early 20th century by Frederic Kipping.[14]
Starting in the 1920s, the work of William Lawrence Bragg on X-ray crystallography elucidated the compositions of the silicates, which had previously been known from analytical chemistry but had not yet been understood, together with Linus Pauling’s development of crystal chemistry and Victor Goldschmidt’s development of geochemistry. The middle of the 20th century saw the development of the chemistry and industrial use of siloxanes and the growing use of silicone polymers, elastomers, and resins. In the late 20th century, the complexity of the crystal chemistry of silicides was mapped, along with the solid-state physics of doped semiconductors.[14]
Silicon semiconductors[edit]
The first semiconductor devices did not use silicon, but used galena, including German physicist Ferdinand Braun’s crystal detector in 1874 and Indian physicist Jagadish Chandra Bose’s radio crystal detector in 1901.[31][32] The first silicon semiconductor device was a silicon radio crystal detector, developed by American engineer Greenleaf Whittier Pickard in 1906.[32]
In 1940, Russell Ohl discovered the p–n junction and photovoltaic effects in silicon. In 1941, techniques for producing high-purity germanium and silicon crystals were developed for radar microwave detector crystals during World War II.[31] In 1947, physicist William Shockley theorized a field-effect amplifier made from germanium and silicon, but he failed to build a working device, before eventually working with germanium instead. The first working transistor was a point-contact transistor built by John Bardeen and Walter Brattain later that year while working under Shockley.[33] In 1954, physical chemist Morris Tanenbaum fabricated the first silicon junction transistor at Bell Labs.[34] In 1955, Carl Frosch and Lincoln Derick at Bell Labs accidentally discovered that silicon dioxide (SiO
2) could be grown on silicon,[35] and they later proposed this could mask silicon surfaces during diffusion processes in 1958.[36]
Silicon Age[edit]
The «Silicon Age» refers to the late 20th century to early 21st century.[37][38][39] This is due to silicon being the dominant material of the Silicon Age (also known as the Digital Age or Information Age), similar to how the Stone Age, Bronze Age and Iron Age were defined by the dominant materials during their respective ages of civilization.[37]
Because silicon is an important element in high-technology semiconductor devices, many places in the world bear its name. For example, Santa Clara Valley in California acquired the nickname Silicon Valley, as the element is the base material in the semiconductor industry there. Since then, many other places have been dubbed similarly, including Silicon Wadi in Israel, Silicon Forest in Oregon, Silicon Hills in Austin, Texas, Silicon Slopes in Salt Lake City, Utah, Silicon Saxony in Germany, Silicon Valley in India, Silicon Border in Mexicali, Mexico, Silicon Fen in Cambridge, England, Silicon Roundabout in London, Silicon Glen in Scotland, Silicon Gorge in Bristol, England, Silicon Alley in New York, New York and Silicon Beach in Los Angeles, California.[40]
Characteristics[edit]
Physical and atomic[edit]
A silicon atom has fourteen electrons. In the ground state, they are arranged in the electron configuration [Ne]3s23p2. Of these, four are valence electrons, occupying the 3s orbital and two of the 3p orbitals. Like the other members of its group, the lighter carbon and the heavier germanium, tin, and lead, it has the same number of valence electrons as valence orbitals: hence, it can complete its octet and obtain the stable noble gas configuration of argon by forming sp3 hybrid orbitals, forming tetrahedral SiX
4 derivatives where the central silicon atom shares an electron pair with each of the four atoms it is bonded to.[42] The first four ionisation energies of silicon are 786.3, 1576.5, 3228.3, and 4354.4 kJ/mol respectively; these figures are high enough to preclude the possibility of simple cationic chemistry for the element. Following periodic trends, its single-bond covalent radius of 117.6 pm is intermediate between those of carbon (77.2 pm) and germanium (122.3 pm). The hexacoordinate ionic radius of silicon may be considered to be 40 pm, although this must be taken as a purely notional figure given the lack of a simple Si4+
cation in reality.[43]
Electrical[edit]
At standard temperature and pressure, silicon is a shiny semiconductor with a bluish-grey metallic lustre; as typical for semiconductors, its resistivity drops as temperature rises. This arises because silicon has a small energy gap (band gap) between its highest occupied energy levels (the valence band) and the lowest unoccupied ones (the conduction band). The Fermi level is about halfway between the valence and conduction bands and is the energy at which a state is as likely to be occupied by an electron as not. Hence pure silicon is effectively an insulator at room temperature. However, doping silicon with a pnictogen such as phosphorus, arsenic, or antimony introduces one extra electron per dopant and these may then be excited into the conduction band either thermally or photolytically, creating an n-type semiconductor. Similarly, doping silicon with a group 13 element such as boron, aluminium, or gallium results in the introduction of acceptor levels that trap electrons that may be excited from the filled valence band, creating a p-type semiconductor.[44] Joining n-type silicon to p-type silicon creates a p–n junction with a common Fermi level; electrons flow from n to p, while holes flow from p to n, creating a voltage drop. This p–n junction thus acts as a diode that can rectify alternating current that allows current to pass more easily one way than the other. A transistor is an n–p–n junction, with a thin layer of weakly p-type silicon between two n-type regions. Biasing the emitter through a small forward voltage and the collector through a large reverse voltage allows the transistor to act as a triode amplifier.[44]
Crystal structure[edit]
Silicon crystallises in a giant covalent structure at standard conditions, specifically in a diamond cubic lattice (space group 227). It thus has a high melting point of 1414 °C, as a lot of energy is required to break the strong covalent bonds and melt the solid. Upon melting silicon contracts as the long-range tetrahedral network of bonds breaks up and the voids in that network are filled in, similar to water ice when hydrogen bonds are broken upon melting. It does not have any thermodynamically stable allotropes at standard pressure, but several other crystal structures are known at higher pressures. The general trend is one of increasing coordination number with pressure, culminating in a hexagonal close-packed allotrope at about 40 gigapascals known as Si–VII (the standard modification being Si–I). An allotrope called BC8 (or bc8), having a body-centred cubic lattice with eight atoms per primitive unit cell (space group 206), can be created at high pressure and remains metastable at low pressure. Its properties have been studied in detail.[45]
Silicon boils at 3265 °C: this, while high, is still lower than the temperature at which its lighter congener carbon sublimes (3642 °C) and silicon similarly has a lower heat of vaporisation than carbon, consistent with the fact that the Si–Si bond is weaker than the C–C bond.[44]
It is also possible to construct silicene layers analogous to graphene.[27][28]
Isotopes[edit]
Naturally occurring silicon is composed of three stable isotopes, 28Si (92.23%), 29Si (4.67%), and 30Si (3.10%).[46] Out of these, only 29Si is of use in NMR and EPR spectroscopy,[47] as it is the only one with a nuclear spin (I =1/2).[29] All three are produced in Type Ia supernovae[48][49] through the oxygen-burning process, with 28Si being made as part of the alpha process and hence the most abundant. The fusion of 28Si with alpha particles by photodisintegration rearrangement in stars is known as the silicon-burning process; it is the last stage of stellar nucleosynthesis before the rapid collapse and violent explosion of the star in question in a type II supernova.[50]
Twenty radioisotopes have been characterized, the two stablest being 32Si with a half-life of about 150 years, and 31Si with a half-life of 2.62 hours.[46] All the remaining radioactive isotopes have half-lives that are less than seven seconds, and the majority of these have half-lives that are less than one tenth of a second.[46] Silicon has one known nuclear isomer, 34mSi, with a half-life less than 210 nanoseconds.[46] 32Si undergoes low-energy beta decay to 32P and then stable 32S. 31Si may be produced by the neutron activation of natural silicon and is thus useful for quantitative analysis; it can be easily detected by its characteristic beta decay to stable 31P, in which the emitted electron carries up to 1.48 MeV of energy.[29]
The known isotopes of silicon range in mass number from 22 to 44.[46] The most common decay mode of the isotopes with mass numbers lower than the three stable isotopes is inverse beta decay, primarily forming aluminium isotopes (13 protons) as decay products.[46] The most common decay mode for the heavier unstable isotopes is beta decay, primarily forming phosphorus isotopes (15 protons) as decay products.[46]
Silicon can enter the oceans through groundwater and riverine transport. Large fluxes of groundwater input have an isotopic composition which is distinct from riverine silicon inputs. Isotopic variations in groundwater and riverine transports contribute to variations in oceanic 30Si values. Currently, there are substantial differences in the isotopic values of deep water in the world’s ocean basins. Between the Atlantic and Pacific oceans, there is a deep water 30Si gradient of greater than 0.3 parts per thousand. 30Si is most commonly associated with productivity in the oceans.[51]
Chemistry and compounds[edit]
| X = | C | Si | H | F | Cl | Br | I | O– | N< |
|---|---|---|---|---|---|---|---|---|---|
| C–X | 368 | 360 | 435 | 453 | 351 | 293 | 216 | ~360 | ~305 |
| Si–X | 360 | 340 | 393 | 565 | 381 | 310 | 234 | 452 | 322 |
Crystalline bulk silicon is rather inert, but becomes more reactive at high temperatures. Like its neighbour aluminium, silicon forms a thin, continuous surface layer of silicon dioxide (SiO
2) that protects the metal from oxidation. Thus silicon does not measurably react with the air below 900 °C, but formation of the vitreous dioxide rapidly increases between 950 °C and 1160 °C and when 1400 °C is reached, atmospheric nitrogen also reacts to give the nitrides SiN and Si
3N
4. Silicon reacts with gaseous sulfur at 600 °C and gaseous phosphorus at 1000 °C. This oxide layer nevertheless does not prevent reaction with the halogens; fluorine attacks silicon vigorously at room temperature, chlorine does so at about 300 °C, and bromine and iodine at about 500 °C. Silicon does not react with most aqueous acids, but is oxidised and complexed by hydrofluoric acid mixtures containing either chlorine or nitric acid to form hexafluorosilicates. It readily dissolves in hot aqueous alkali to form silicates.[52] At high temperatures, silicon also reacts with alkyl halides; this reaction may be catalysed by copper to directly synthesise organosilicon chlorides as precursors to silicone polymers. Upon melting, silicon becomes extremely reactive, alloying with most metals to form silicides, and reducing most metal oxides because the heat of formation of silicon dioxide is so large. In fact, molten silicon reacts virtually with every known kind of crucible material (except its own oxide, SiO
2).[53]: 13 This happens due to silicon’s high binding forces for the light elements and to its high dissolving power for most elements.[53]: 13 As a result, containers for liquid silicon must be made of refractory, unreactive materials such as zirconium dioxide or group 4, 5, and 6 borides.[44][54]
Tetrahedral coordination is a major structural motif in silicon chemistry just as it is for carbon chemistry. However, the 3p subshell is rather more diffuse than the 2p subshell and does not hybridise so well with the 3s subshell. As a result, the chemistry of silicon and its heavier congeners shows significant differences from that of carbon,[55] and thus octahedral coordination is also significant.[44] For example, the electronegativity of silicon (1.90) is much less than that of carbon (2.55), because the valence electrons of silicon are further from the nucleus than those of carbon and hence experience smaller electrostatic forces of attraction from the nucleus. The poor overlap of 3p orbitals also results in a much lower tendency toward catenation (formation of Si–Si bonds) for silicon than for carbon, due to the concomitant weakening of the Si–Si bond compared to the C–C bond:[56] the average Si–Si bond energy is approximately 226 kJ/mol, compared to a value of 356 kJ/mol for the C–C bond.[57] This results in multiply bonded silicon compounds generally being much less stable than their carbon counterparts, an example of the double bond rule. On the other hand, the presence of radial nodes in the 3p orbitals of silicon suggests the possibility of hypervalence, as seen in five and six-coordinate derivatives of silicon such as SiX−
5 and SiF2−
6.[58][56] Lastly, because of the increasing energy gap between the valence s and p orbitals as the group is descended, the divalent state grows in importance from carbon to lead, so that a few unstable divalent compounds are known for silicon; this lowering of the main oxidation state, in tandem with increasing atomic radii, results in an increase of metallic character down the group. Silicon already shows some incipient metallic behavior, particularly in the behavior of its oxide compounds and its reaction with acids as well as bases (though this takes some effort), and is hence often referred to as a metalloid rather than a nonmetal.[56] However, metallicity does not become clear in group 14 until germanium and dominant until tin, with the growing importance of the lower +2 oxidation state.[14]
Silicon shows clear differences from carbon. For example, organic chemistry has very few analogies with silicon chemistry, while silicate minerals have a structural complexity unseen in oxocarbons.[59] Silicon tends to resemble germanium far more than it does carbon, and this resemblance is enhanced by the d-block contraction, resulting in the size of the germanium atom being much closer to that of the silicon atom than periodic trends would predict.[60] Nevertheless, there are still some differences because of the growing importance of the divalent state in germanium compared to silicon, which result in germanium being significantly more metallic than silicon. Additionally, the lower Ge–O bond strength compared to the Si–O bond strength results in the absence of «germanone» polymers that would be analogous to silicone polymers.[57]
Silicides[edit]
Phase diagram of the Fe–Si system
Many metal silicides are known, most of which have formulae that cannot be explained through simple appeals to valence: their bonding ranges from metallic to ionic and covalent. Some known stoichiometries are M
6Si, M
5Si, M
4Si, M
15Si
4, M
3Si, M
5Si
2, M
2Si, M
5Si
3, M
3Si
2, MSi, M
2Si
3, MSi
2, MSi
3, and MSi
6. They are structurally more similar to the borides than the carbides, in keeping with the diagonal relationship between boron and silicon, although the larger size of silicon than boron means that exact structural analogies are few and far between. The heats of formation of the silicides are usually similar to those of the borides and carbides of the same elements, but they usually melt at lower temperatures.[61] Silicides are known for all stable elements in groups 1–10, with the exception of beryllium: in particular, uranium and the transition metals of groups 4–10 show the widest range of stoichiometries. Except for copper, the metals in groups 11–15 do not form silicides. Instead, most form eutectic mixtures, although the heaviest post-transition metals mercury, thallium, lead, and bismuth are completely immiscible with liquid silicon.[44]
Usually, silicides are prepared by direct reaction of the elements. For example, the alkali metals and alkaline earth metals react with silicon or silicon oxide to give silicides. Nevertheless, even with these highly electropositive elements true silicon anions are not obtainable, and most of these compounds are semiconductors. For example, the alkali metal silicides (M+
)
4(Si4−
4) contain pyramidal tricoordinate silicon in the Si4−
4 anion, isoelectronic with white phosphorus, P
4.[44][62] Metal-rich silicides tend to have isolated silicon atoms (e. g. Cu
5Si); with increasing silicon content, catenation increases, resulting in isolated clusters of two (e. g. U
3Si
2) or four silicon atoms (e. g. [K+
]
4[Si
4]4−
) at first, followed by chains (e. g. CaSi), layers (e. g. CaSi
2), or three-dimensional networks of silicon atoms spanning space (e. g. α-ThSi
2) as the silicon content rises even higher.[44]
The silicides of the group 1 and 2 metals usually are more reactive than the transition metal silicides. The latter usually do not react with aqueous reagents, except for hydrofluoric acid; however, they do react with much more aggressive reagents such as liquid potassium hydroxide, or gaseous fluorine or chlorine when red-hot. The pre-transition metal silicides instead readily react with water and aqueous acids, usually producing hydrogen or silanes:[44]
- Na
2Si + 3 H2O → Na
2SiO
3 + 3 H
2 - Mg
2Si + 2 H
2SO
4 → 2 MgSO
4 + SiH
4
Products often vary with the stoichiometry of the silicide reactant. For example, Ca
2Si is polar and non-conducting and has the anti-PbCl
2 structure with single isolated silicon atoms, and reacts with water to produce calcium hydroxide, hydrated silicon dioxide, and hydrogen gas. CaSi with its zigzag chains of silicon atoms instead reacts to give silanes and polymeric SiH
2, while CaSi
2 with its puckered layers of silicon atoms does not react with water, but will react with dilute hydrochloric acid: the product is a yellow polymeric solid with stoichiometry Si
2H
2O.[44]
Silanes[edit]
Speculation on silicon hydride chemistry started in the 1830s, contemporary with the development of synthetic organic chemistry. Silane itself, as well as trichlorosilane, were first synthesised by Friedrich Wöhler and Heinrich Buff in 1857 by reacting aluminium–silicon alloys with hydrochloric acid, and characterised as SiH
4 and SiHCl
3 by Charles Friedel and Albert Ladenburg in 1867. Disilane (Si
2H
6) followed in 1902, when it was first made by Henri Moissan and Samuel Smiles by the protonolysis of magnesium silicides. Further investigation had to wait until 1916 because of the great reactivity and thermal instability of the silanes; it was then that Alfred Stock began to study silicon hydrides in earnest with new greaseless vacuum techniques, as they were found as contaminants of his focus, the boron hydrides. The names silanes and boranes are his, based on analogy with the alkanes.[29][63][64] The Moissan and Smiles method of preparation of silanes and silane derivatives via protonolysis of metal silicides is still used, although the yield is lowered by the hydrolysis of the products that occurs simultaneously, so that the preferred route today is to treat substituted silanes with hydride reducing agents such as lithium aluminium hydride in etheric solutions at low temperatures. Direct reaction of HX or RX with silicon, possibly with a catalyst such as copper, is also a viable method of producing substituted silanes.[29]
The silanes comprise a homologous series of silicon hydrides with a general formula of Si
nH
2n + 2. They are all strong reducing agents. Unbranched and branched chains are known up to n=8, and the cycles Si
5H
10 and Si
6H
12 are also known. The first two, silane and disilane, are colourless gases; the heavier members of the series are volatile liquids. All silanes are very reactive and catch fire or explode spontaneously in air. They become less thermally stable with room temperature, so that only silane is indefinitely stable at room temperature, although disilane does not decompose very quickly (only 2.5% of a sample decomposes after the passage of eight months).[29] They decompose to form polymeric polysilicon hydride and hydrogen gas.[65][66] As expected from the difference in atomic weight, the silanes are less volatile than the corresponding alkanes and boranes, but more so than the corresponding germanes. They are much more reactive than the corresponding alkanes, because of the larger radius of silicon compared to carbon facilitating nucleophilic attack at the silicon, the greater polarity of the Si–H bond compared to the C–H bond, and the ability of silicon to expand its octet and hence form adducts and lower the reaction’s activation energy.[29]
Silane pyrolysis gives polymeric species and finally elemental silicon and hydrogen; indeed ultrapure silicon is commercially produced by the pyrolysis of silane. While the thermal decomposition of alkanes starts by the breaking of a C–H or C–C bond and the formation of radical intermediates, polysilanes decompose by eliminating silylenes :SiH
2 or :SiHR, as the activation energy of this process (~210 kJ/mol) is much less than the Si–Si and Si–H bond energies. While pure silanes do not react with pure water or dilute acids, traces of alkali catalyse immediate hydrolysis to hydrated silicon dioxide. If the reaction is carried out in methanol, controlled solvolysis results in the products SiH
2(OMe)
2, SiH(OMe)
3, and Si(OMe)
4. The Si–H bond also adds to alkenes, a reaction which proceeds slowly and speeds up with increasing substitution of the silane involved. At 450 °C, silane participates in an addition reaction with acetone, as well as a ring-opening reaction with ethylene oxide. Direct reaction of the silanes with chlorine or bromine results in explosions at room temperature, but the reaction of silane with bromine at −80 °C is controlled and yields bromosilane and dibromosilane. The monohalosilanes may be formed by reacting silane with the appropriate hydrogen halide with an Al
2X
6 catalyst, or by reacting silane with a solid silver halide in a heated flow reactor:[29]
- SiH
4 + 2 AgCl 260 °C→ SiH
3Cl + HCl + 2 Ag
Among the derivatives of silane, iodosilane (SiH
3I) and potassium silanide (KSiH
3) are very useful synthetic intermediates in the production of more complicated silicon-containing compounds: the latter is a colourless crystalline ionic solid containing K+ cations and SiH−
3 anions in the NaCl structure, and is made by the reduction of silane by potassium metal.[67] Additionally, the reactive hypervalent species SiH−
5 is also known.[29] With suitable organic substituents it is possible to produce stable polysilanes: they have surprisingly high electric conductivities, arising from sigma delocalisation of the electrons in the chain.[68]
Halides[edit]
Silicon and silicon carbide readily react with all four stable halogens, forming the colourless, reactive, and volatile silicon tetrahalides[69] Silicon tetrafluoride also may be made by fluorinating the other silicon halides, and is produced by the attack of hydrofluoric acid on glass.[70] Heating two different tetrahalides together also produces a random mixture of mixed halides, which may also be produced by halogen exchange reactions. The melting and boiling points of these species usually rise with increasing atomic weight, though there are many exceptions: for example, the melting and boiling points drop as one passes from SiFBr
3 through SiFClBr
2 to SiFCl
2Br. The shift from the hypoelectronic elements in Group 13 and earlier to the Group 14 elements is illustrated by the change from an infinite ionic structure in aluminium fluoride to a lattice of simple covalent silicon tetrafluoride molecules, as dictated by the lower electronegativity of aluminium than silicon, the stoichiometry (the +4 oxidation state being too high for true ionicity), and the smaller size of the silicon atom compared to the aluminium atom.[69]
Silicon tetrachloride is manufactured on a huge scale as a precursor to the production of pure silicon, silicon dioxide, and some silicon esters.[69] The silicon tetrahalides hydrolyse readily in water, unlike the carbon tetrahalides, again because of the larger size of the silicon atom rendering it more open to nucleophilic attack and the ability of the silicon atom to expand its octet which carbon lacks.[70] The reaction of silicon tetrafluoride with excess hydrofluoric acid produces the octahedral hexafluorosilicate anion SiF2−
6.[70]
Analogous to the silanes, halopolysilanes Si
nX
2n + 2 also are known. While catenation in carbon compounds is maximised in the hydrogen compounds rather than the halides, the opposite is true for silicon, so that the halopolysilanes are known up to at least Si
14F
30, Si
6Cl
14, and Si
4Br
10. A suggested explanation for this phenomenon is the compensation for the electron loss of silicon to the more electronegative halogen atoms by pi backbonding from the filled pπ orbitals on the halogen atoms to the empty dπ orbitals on silicon: this is similar to the situation of carbon monoxide in metal carbonyl complexes and explains their stability. These halopolysilanes may be produced by comproportionation of silicon tetrahalides with elemental silicon, or by condensation of lighter halopolysilanes (trimethylammonium being a useful catalyst for this reaction).[69]
Silica[edit]
Silicon dioxide (SiO
2), also known as silica, is one of the best-studied compounds, second only to water. Twelve different crystal modifications of silica are known, the most common being α-quartz, a major constituent of many rocks such as granite and sandstone. It also is known to occur in a pure form as rock crystal; impure forms are known as rose quartz, smoky quartz, morion, amethyst, and citrine. Some poorly crystalline forms of quartz are also known, such as chalcedony, chrysoprase, carnelian, agate, onyx, jasper, heliotrope, and flint. Other modifications of silicon dioxide are known in some other minerals such as tridymite and cristobalite, as well as the much less common coesite and stishovite. Biologically generated forms are also known as kieselguhr and diatomaceous earth. Vitreous silicon dioxide is known as tektites, and obsidian, and rarely as lechatelierite. Some synthetic forms are known as keatite. Opals are composed of complicated crystalline aggregates of partially hydrated silicon dioxide.[71]
-
Quartz
-
Agate
-
Tridymite
-
Cristobalite
-
Coesite
Most crystalline forms of silica are made of infinite arrangements of SiO tetrahedra (with Si at the center) connected at their corners, with each oxygen atom linked to two silicon atoms. In the thermodynamically stable room-temperature form, α-quartz, these tetrahedra are linked in intertwined helical chains with two different Si–O distances (159.7 and 161.7 pm) with a Si–O–Si angle of 144°. These helices can be either left- or right-handed, so that individual α-quartz crystals are optically active. At 537 °C, this transforms quickly and reversibly into the similar β-quartz, with a change of the Si–O–Si angle to 155° but a retention of handedness. Further heating to 867 °C results in another reversible phase transition to β-tridymite, in which some Si–O bonds are broken to allow for the arrangement of the SiO tetrahedra into a more open and less dense hexagonal structure. This transition is slow and hence tridymite occurs as a metastable mineral even below this transition temperature; when cooled to about 120 °C it quickly and reversibly transforms by slight displacements of individual silicon and oxygen atoms to α-tridymite, similarly to the transition from α-quartz to β-quartz. β-tridymite slowly transforms to cubic β-cristobalite at about 1470 °C, which once again exists metastably below this transition temperature and transforms at 200–280 °C to α-cristobalite via small atomic displacements. β-cristobalite melts at 1713 °C; the freezing of silica from the melt is quite slow and vitrification, or the formation of a glass, is likely to occur instead. In vitreous silica, the SiO tetrahedra remain corner-connected, but the symmetry and periodicity of the crystalline forms are lost. Because of the slow conversions between these three forms, it is possible upon rapid heating to melt β-quartz (1550 °C) or β-tridymite (1703 °C). Silica boils at approximately 2800 °C. Other high-pressure forms of silica are known, such as coesite and stishovite: these are known in nature, formed under the shock pressure of a meteorite impact and then rapidly quenched to preserve the crystal structure. Similar melting and cooling of silica occurs following lightning strikes, forming glassy lechatelierite. W-silica is an unstable low-density form involving SiO tetrahedra sharing opposite edges instead of corners, forming parallel chains similarly to silicon disulfide (SiS
2) and silicon diselenide (SiSe
2): it quickly returns to forming amorphous silica with heat or traces of water[72]
Condensed polysilicic acid
Silica is rather inert chemically. It is not attacked by any acids other than hydrofluoric acid. However, it slowly dissolves in hot concentrated alkalis, and does so rather quickly in fused metal hydroxides or carbonates, to give metal silicates. Among the elements, it is attacked only by fluorine at room temperature to form silicon tetrafluoride: hydrogen and carbon also react, but require temperatures over 1000 °C to do so. Silica nevertheless reacts with many metal and metalloid oxides to form a wide variety of compounds important in the glass and ceramic industries above all, but also have many other uses: for example, sodium silicate is often used in detergents due to its buffering, saponifying, and emulsifying properties[72]
Silicic acids[edit]
Adding water to silica drops its melting point by around 800 °C due to the breaking of the structure by replacing Si–O–Si linkages with terminating Si–OH groups. Increasing water concentration results in the formation of hydrated silica gels and colloidal silica dispersions. Many hydrates and silicic acids exist in the most dilute of aqueous solutions, but these are rather insoluble and quickly precipitate and condense and cross-link to form various polysilicic acids of variable combinations following the formula [SiO
x(OH)
4−2x]
n, similar to the behaviour of boron, aluminium, and iron, among other elements. Hence, although some simple silicic acids have been identified in dilute solutions, such as orthosilicic acid Si(OH)
4 and metasilicic acid SiO(OH)
2, none of these are likely to exist in the solid state.[72]
Silicate minerals[edit]
| CN 4 | LiI (59) |
BeII (27) | AlIII (39) | SiIV (26) | |
|---|---|---|---|---|---|
| CN 6 | NaI (102) | MgII (72) | AlIII (54) | TiIV (61) | FeII (78) |
| CN 8 | KI (151) | CaII (112) | |||
| CN 12 | KI (164) |
About 95% of the Earth’s crustal rocks are made of silica or silicate and aluminosilicate minerals, as reflected in oxygen, silicon, and aluminium being the three most common elements in the crust (in that order).[73] Measured by mass, silicon makes up 27.7% of the Earth’s crust.[74] Pure silicon crystals are very rarely found in nature, but notable exceptions are crystals as large as to 0.3 mm across found during sampling gases from the Kudriavy volcano on Iturup, one of the Kuril Islands.[75][76]
Silicate and aluminosilicate minerals have many different structures and varying stoichiometry, but they may be classified following some general principles. Tetrahedral SiO units are common to almost all these compounds, either as discrete structures, or combined into larger units by the sharing of corner oxygen atoms. These may be divided into neso-silicates (discrete SiO units) sharing no oxygen atoms, soro-silicates (discrete Si units) sharing one, cyclo-silicates (closed ring structures) and ino-silicates (continuous chain or ribbon structures) both sharing two, phyllo-silicates (continuous sheets) sharing three, and tecto-silicates (continuous three-dimensional frameworks) sharing four. The lattice of oxygen atoms that results is usually close-packed, or close to it, with the charge being balanced by other cations in various different polyhedral sites according to size.[77]
The orthosilicates MII
2SiO
4 (M = Be, Mg, Mn, Fe, Zn) and ZrSiO
4 are neso-silicates. Be
2SiO
4 (phenacite) is unusual as both BeII and SiIV occupy tetrahedral four-coordinated sites; the other divalent cations instead occupy six-coordinated octahedral sites and often isomorphously replace each other as in olivine, (Mg,Fe,Mn)
2SiO
4. Zircon, ZrSiO
4, demands eight-coordination of the ZrIV cations due to stoichiometry and because of their larger ionic radius (84 pm). Also significant are the garnets, [MII
3MIII
2(SiO
4)
3], in which the divalent cations (e.g. Ca, Mg, Fe) are eight-coordinated and the trivalent ones are six-coordinated (e.g. Al, Cr, Fe). Regular coordination is not always present: for example, it is not found in Ca
2SiO
4, which mixes six- and eight-coordinate sites for CaII. Soro-silicates, involving discrete double or triple tetrahedral units, are quite rare: metasilicates involving cyclic «[(SiOn
3)]2n−» units of corner-abutting tetrahedra forming a polygonal ring are also known.[73]
Chain metasilicates, {SiO2−
3}
∞, form by corner-sharing of an indefinite chain of linked SiO tetrahedra. Many differences arise due to the differing repeat distances of conformation across the line of tetrahedra. A repeat distance of two is most common, as in most pyroxene minerals, but repeat distances of one, three, four, five, six, seven, nine, and twelve are also known. These chains may then link across each other to form double chains and ribbons, as in the asbestos minerals, involving repeated chains of cyclic tetrahedron rings.[73]
A typical zeolite structure
Layer silicates, such as the clay minerals and the micas, are very common, and often are formed by horizontal cross-linking of metasilicate chains or planar condensation of smaller units. An example is kaolinite [Al
2(OH)
4Si
2O
5]; in many of these minerals cation and anion replacement is common, so that for example tetrahedral SiIV may be replaced by AlIII, octahedral AlIII by MgII, and OH−
by F−
. Three-dimensional framework aluminosilicates are structurally very complex; they may be conceived of as starting from the SiO
2 structure, but having replaced up to one-half of the SiIV atoms with AlIII, they require more cations to be included in the structure to balance charge. Examples include feldspars (the most abundant minerals on the Earth), zeolites, and ultramarines. Many feldspars can be thought of as forming part of the ternary system NaAlSi
3O
8–KAlSi
3O
8–CaAl
2Si
2O
8. Their lattice is destroyed by high pressure prompting AlIII to undergo six-coordination rather than four-coordination, and this reaction destroying feldspars may be a reason for the Mohorovičić discontinuity, which would imply that the crust and mantle have the same chemical composition, but different lattices, although this is not a universally held view. Zeolites have many polyhedral cavities in their frameworks (truncated cuboctahedra being most common, but other polyhedra also are known as zeolite cavities), allowing them to include loosely bound molecules such as water in their structure. Ultramarines alternate silicon and aluminium atoms and include a variety of other anions such as Cl−, SO2−
4, and S2−
2, but are otherwise similar to the feldspars.[73]
Other inorganic compounds[edit]
Silicon disulfide (SiS
2) is formed by burning silicon in gaseous sulfur at 100 °C; sublimation of the resulting compound in nitrogen results in white, flexible long fibers reminiscent of asbestos with a structure similar to W-silica. This melts at 1090 °C and sublimes at 1250 °C; at high temperature and pressure this transforms to a crystal structure analogous to cristobalite. However, SiS
2 lacks the variety of structures of SiO
2, and quickly hydrolyses to silica and hydrogen sulfide. It is also ammonolysed quickly and completely by liquid ammonia as follows to form an imide:[60]
- SiS
2 + 4 NH
3 → Si(NH)
2 + 2 NH
4SH
It reacts with the sulfides of sodium, magnesium, aluminium, and iron to form metal thiosilicates: reaction with ethanol results in tetraethylsilicate Si(OEt)
4 and hydrogen sulfide. Ethylsilicate is useful as its controlled hydrolysis produces adhesive or film-like forms of silica. Reacting hydrogen sulfide with silicon tetrahalides yields silicon thiohalides such as S(SiCl)
3, cyclic Cl
2Si(μ-S)
2SiCl
2, and crystalline (SiSCl
2)
4. Despite the double bond rule, stable organosilanethiones RR’Si=S have been made thanks to the stabilising mechanism of intermolecular coordination via an amine group.[78]
Silicon nitride, Si
3N
4, may be formed by directly reacting silicon with nitrogen above 1300 °C, but a more economical means of production is by heating silica and coke in a stream of nitrogen and hydrogen gas at 1500 °C. It would make a promising ceramic if not for the difficulty of working with and sintering it: chemically, it is near-totally inert, and even above 1000 °C it keeps its strength, shape, and continues to be resistant to wear and corrosion. It is very hard (9 on the Mohs hardness scale), dissociates only at 1900 °C at 1 atm, and is quite dense (density 3.185 g/cm3), because of its compact structure similar to that of phenacite (Be
2SiO
4). A similar refractory material is Si
2N
2O, formed by heating silicon and silica at 1450 °C in an argon stream containing 5% nitrogen gas, involving 4-coordinate silicon and 3-coordinate nitrogen alternating in puckered hexagonal tilings interlinked by non-linear Si–O–Si linkages to each other.[78]
Reacting silyl halides with ammonia or alkylammonia derivatives in the gaseous phase or in ethanolic solution produces various volatile silylamides, which are silicon analogues of the amines:[78]
- 3 SiH
3Cl + 4 NH
3 → N(SiH
3)
3 + 3 NH
4Cl - SiH
3Br + 2 Me
2NH → SiH
3NMe
2 + Me
2NH
2Br - 4 SiH
3I + 5 N
2H
4 → (SiH
3)
2NN(SiH
3)
2 + 4 N
2H
5I
Many such compounds have been prepared, the only known restriction being that the nitrogen is always tertiary, and species containing the SiH–NH group are unstable at room temperature. The stoichiometry around the nitrogen atom in compounds such as N(SiH
3)
3 is planar. Similarly, trisilylamines are weaker as ligands than their carbon analogues, the tertiary amines, although substitution of some SiH
3 groups by CH
3 groups mitigates this weakness. For example, N(SiH
3)
3 does not form an adduct with BH
3 at all, while MeN(SiH
3)
2 and Me
2NSiH
3 form adducts at low temperatures that decompose upon warming. Some silicon analogues of imines, with a Si=N double bond, are known: the first found was But2Si=N–SiBut3, which was discovered in 1986.[78]
Silicon carbide (SiC) was first made by Edward Goodrich Acheson in 1891, who named it carborundum to reference its intermediate hardness and abrasive power between diamond (an allotrope of carbon) and corundum (aluminium oxide). He soon founded a company to manufacture it, and today about one million tonnes are produced each year.[79] Silicon carbide exists in about 250 crystalline forms.[80] The polymorphism of SiC is characterized by a large family of similar crystalline structures called polytypes. They are variations of the same chemical compound that are identical in two dimensions and differ in the third. Thus they can be viewed as layers stacked in a certain sequence.[81] It is made industrially by reduction of quartz sand with excess coke or anthracite at 2000–2500 °C in an electric furnace:[79]
- SiO
2 + 2 C → Si + 2 CO - Si + C → SiC
It is the most thermally stable binary silicon compound, only decomposing through loss of silicon starting from around 2700 °C. It is resistant to most aqueous acids, phosphoric acid being an exception. It forms a protective layer of silicon dioxide on the surface and hence only oxidises appreciably in air above 1000 °C; removal of this layer by molten hydroxides or carbonates leads to quick oxidation. Silicon carbide is rapidly attacked by chlorine gas, which forms SiCl
4 and carbon at 100 °C and SiCl
4 and CCl
4 at 1000 °C. It is mostly used as an abrasive and a refractory material, as it is chemically stable and very strong, and it fractures to form a very sharp cutting edge. It is also useful as an intrinsic semiconductor, as well as an extrinsic semiconductor upon being doped.[79] In its diamond-like behavior it serves as an illustration of the chemical similarity between carbon and silicon.[82]
Organosilicon compounds[edit]
A hydrosilylation reaction, in which Si–H is added to an unsaturated substrate
Because the Si–C bond is close in strength to the C–C bond, organosilicon compounds tend to be markedly thermally and chemically stable. For example, tetraphenylsilane (SiPh
4) may be distilled in air even at its boiling point of 428 °C, and so may its substituted derivatives Ph
3SiCl and Ph
2SiCl
2, which boil at 378 °C and 305 °C respectively. Furthermore, since carbon and silicon are chemical congeners, organosilicon chemistry shows some significant similarities with carbon chemistry, for example in the propensity of such compounds for catenation and forming multiple bonds.[82] However, significant differences also arise: since silicon is more electropositive than carbon, bonds to more electronegative elements are generally stronger with silicon than with carbon, and vice versa. Thus the Si–F bond is significantly stronger than even the C–F bond and is one of the strongest single bonds, while the Si–H bond is much weaker than the C–H bond and is readily broken. Furthermore, the ability of silicon to expand its octet is not shared by carbon, and hence some organosilicon reactions have no organic analogues. For example, nucleophilic attack on silicon does not proceed by the SN2 or SN1 processes, but instead goes through a negatively charged true pentacoordinate intermediate and appears like a substitution at a hindered tertiary atom. This works for silicon, unlike for carbon, because the long Si–C bonds reduce the steric hindrance and there are no geometric constraints for nucleophilic attack, unlike for example a C–O σ* antibonding orbital. Nevertheless, despite these differences, the mechanism is still often called «SN2 at silicon» for simplicity.[83]
One of the most useful silicon-containing groups is trimethylsilyl, Me
3Si–. The Si–C bond connecting it to the rest of the molecule is reasonably strong, allowing it to remain while the rest of the molecule undergoes reactions, but is not so strong that it cannot be removed specifically when needed, for example by the fluoride ion, which is a very weak nucleophile for carbon compounds but a very strong one for organosilicon compounds. It may be compared to acidic protons; while trisilylmethyl is removed by hard nucleophiles instead of bases, both removals usually promote elimination. As a general rule, while saturated carbon is best attacked by nucleophiles that are neutral compounds, those based on nonmetals far down on the periodic table (e.g. sulfur, selenium, or iodine), or even both, silicon is best attacked by charged nucleophiles, particularly those involving such highly electronegative nonmetals as oxygen, fluorine, or chlorine. For example, enolates react at the carbon in haloalkanes, but at the oxygen in silyl chlorides; and when trimethylsilyl is removed from an organic molecule using hydroxide as a nucleophile, the product of the reaction is not the silanol as one would expect from using carbon chemistry as an analogy, because the siloxide is strongly nucleophilic and attacks the original molecule to yield the silyl ether hexamethyldisiloxane, (Me
3Si)
2O. Conversely, while the SN2 reaction is mostly unaffected by the presence of a partial positive charge (δ+) at the carbon, the analogous «SN2″ reaction at silicon is so affected. Thus, for example, the silyl triflates are so electrophilic that they react 108 to 109 times faster than silyl chlorides with oxygen-containing nucleophiles. Trimethylsilyl triflate is in particular a very good Lewis acid and is used to convert carbonyl compounds to acetals and silyl enol ethers, reacting them together analogously to the aldol reaction.[83]
Si–C bonds are commonly formed in three ways. In the laboratory, preparation is often carried out in small quantities by reacting tetrachlorosilane (silicon tetrachloride) with organolithium, Grignard, or organoaluminium reagents, or by catalytic addition of Si–H across C=C double bonds. The second route has the drawback of not being applicable to the most important silanes, the methyl and phenyl silanes. Organosilanes are made industrially by directly reacting alkyl or aryl halides with silicon with 10% by weight metallic copper as a catalyst. Standard organic reactions suffice to produce many derivatives; the resulting organosilanes are often significantly more reactive than their carbon congeners, readily undergoing hydrolysis, ammonolysis, alcoholysis, and condensation to form cyclic oligomers or linear polymers.[82]
Silicone polymers[edit]
The word «silicone» was first used by Frederic Kipping in 1901. He invented the word to illustrate the similarity of chemical formulae between Ph
2SiO and benzophenone, Ph
2CO, although he also stressed the lack of chemical resemblance due to the polymeric structure of Ph
2SiO, which is not shared by Ph
2CO.[82]
Silicones may be considered analogous to mineral silicates, in which the methyl groups of the silicones correspond to the isoelectronic <O−
of the silicates.[82] They are quite stable to extreme temperatures, oxidation, and water, and have useful dielectric, antistick, and antifoam properties. Furthermore, they are resistant over long periods of time to ultraviolet radiation and weathering, and are inert physiologically. They are fairly unreactive, but do react with concentrated solutions bearing the hydroxide ion and fluorinating agents, and occasionally, may even be used as mild reagents for selective syntheses. For example, (Me
3Si)
2O is valuable for the preparation of derivatives of molybdenum and tungsten oxyhalides, converting a tungsten hexachloride suspension in dichloroethane solution quantitatively to WOCl
4 in under an hour at room temperature, and then to yellow WO
2Cl
2 at 100 °C in light petroleum at a yield of 95% overnight.[84]
Occurrence[edit]
Silicon is the eighth most abundant element in the universe, coming after hydrogen, helium, carbon, nitrogen, oxygen, iron, and neon. These abundances are not replicated well on Earth due to substantial separation of the elements taking place during the formation of the Solar System. Silicon makes up 27.2% of the Earth’s crust by weight, second only to oxygen at 45.5%, with which it always is associated in nature. Further fractionation took place in the formation of the Earth by planetary differentiation: Earth’s core, which makes up 31.5% of the mass of the Earth, has approximate composition Fe
25Ni
2Co
0.1S
3; the mantle makes up 68.1% of the Earth’s mass and is composed mostly of denser oxides and silicates, an example being olivine, (Mg,Fe)
2SiO
4; while the lighter siliceous minerals such as aluminosilicates rise to the surface and form the crust, making up 0.4% of the Earth’s mass.[85][86]
The crystallisation of igneous rocks from magma depends on a number of factors; among them are the chemical composition of the magma, the cooling rate, and some properties of the individual minerals to be formed, such as lattice energy, melting point, and complexity of their crystal structure. As magma is cooled, olivine appears first, followed by pyroxene, amphibole, biotite mica, orthoclase feldspar, muscovite mica, quartz, zeolites, and finally, hydrothermal minerals. This sequence shows a trend toward increasingly complex silicate units with cooling, and the introduction of hydroxide and fluoride anions in addition to oxides. Many metals may substitute for silicon. After these igneous rocks undergo weathering, transport, and deposition, sedimentary rocks like clay, shale, and sandstone are formed. Metamorphism also may occur at high temperatures and pressures, creating an even vaster variety of minerals.[85]
There are four sources for silicon fluxes into the ocean include chemical weathering of continental rocks, river transport, dissolution of continental terrigenous silicates, and through the reaction between submarine basalts and hydrothermal fluid which release dissolved silicon. All four of these fluxes are interconnected in the ocean’s biogeochemical cycle as they all were initially formed from the weathering of Earth’s crust.[87]
Approximately 300–900 megatonnes of Aeolian dust is deposited into the world’s oceans each year. Of that value, 80–240 megatonnes are in the form of particulate silicon. The total amount of particulate silicon deposition into the ocean is still less than the amount of silicon influx into the ocean via riverine transportation.[88] Aeolian inputs of particulate lithogenic silicon into the North Atlantic and Western North Pacific oceans are the result of dust settling on the oceans from the Sahara and Gobi Desert, respectively.[87] Riverine transports are the major source of silicon influx into the ocean in coastal regions, while silicon deposition in the open ocean is greatly influenced by the settling of Aeolian dust.[88]
Production[edit]
Silicon of 96–99% purity is made by reducing quartzite or sand with highly pure coke. The reduction is carried out in an electric arc furnace, with an excess of SiO
2 used to stop silicon carbide (SiC) from accumulating:[29]
- SiO
2 + 2 C → Si + 2 CO - 2 SiC + SiO
2 → 3 Si + 2 CO
This reaction, known as carbothermal reduction of silicon dioxide, usually is conducted in the presence of scrap iron with low amounts of phosphorus and sulfur, producing ferrosilicon.[29] Ferrosilicon, an iron-silicon alloy that contains varying ratios of elemental silicon and iron, accounts for about 80% of the world’s production of elemental silicon, with China, the leading supplier of elemental silicon, providing 4.6 million tonnes (or 2/3rds of world output) of silicon, most of it in the form of ferrosilicon. It is followed by Russia (610,000 t), Norway (330,000 t), Brazil (240,000 t), and the United States (170,000 t).[89] Ferrosilicon is primarily used by the iron and steel industry (see below) with primary use as alloying addition in iron or steel and for de-oxidation of steel in integrated steel plants.[29]
Another reaction, sometimes used, is aluminothermal reduction of silicon dioxide, as follows:[90]
- 3 SiO
2 + 4 Al → 3 Si + 2 Al
2O
3
Leaching powdered 96–97% pure silicon with water results in ~98.5% pure silicon, which is used in the chemical industry. However, even greater purity is needed for semiconductor applications, and this is produced from the reduction of tetrachlorosilane (silicon tetrachloride) or trichlorosilane. The former is made by chlorinating scrap silicon and the latter is a byproduct of silicone production. These compounds are volatile and hence can be purified by repeated fractional distillation, followed by reduction to elemental silicon with very pure zinc metal as the reducing agent. The spongy pieces of silicon thus produced are melted and then grown to form cylindrical single crystals, before being purified by zone refining. Other routes use the thermal decomposition of silane or tetraiodosilane (SiI
4). Another process used is the reduction of sodium hexafluorosilicate, a common waste product of the phosphate fertilizer industry, by metallic sodium: this is highly exothermic and hence requires no outside energy source. Hyperfine silicon is made at a higher purity than almost any other material: transistor production requires impurity levels in silicon crystals less than 1 part per 1010, and in special cases impurity levels below 1 part per 1012 are needed and attained.[29]
Silicon nanostructures can directly be produced from silica sand using conventional metalothermic processes, or the combustion synthesis approach. Such nanostructured silicon materials can be used in various functional applications including the anode of lithium ion batteries (LIBs) or phorocatalytic applications.[91]
Applications[edit]
Compounds[edit]
Most silicon is used industrially without being purified, and indeed, often with comparatively little processing from its natural form. More than 90% of the Earth’s crust is composed of silicate minerals, which are compounds of silicon and oxygen, often with metallic ions when negatively charged silicate anions require cations to balance the charge. Many of these have direct commercial uses, such as clays, silica sand, and most kinds of building stone. Thus, the vast majority of uses for silicon are as structural compounds, either as the silicate minerals or silica (crude silicon dioxide). Silicates are used in making Portland cement (made mostly of calcium silicates) which is used in building mortar and modern stucco, but more importantly, combined with silica sand, and gravel (usually containing silicate minerals such as granite), to make the concrete that is the basis of most of the very largest industrial building projects of the modern world.[92]
Silica is used to make fire brick, a type of ceramic. Silicate minerals are also in whiteware ceramics, an important class of products usually containing various types of fired clay minerals (natural aluminium phyllosilicates). An example is porcelain, which is based on the silicate mineral kaolinite. Traditional glass (silica-based soda-lime glass) also functions in many of the same ways, and also is used for windows and containers. In addition, specialty silica based glass fibers are used for optical fiber, as well as to produce fiberglass for structural support and glass wool for thermal insulation.
Silicones often are used in waterproofing treatments, molding compounds, mold-release agents, mechanical seals, high temperature greases and waxes, and caulking compounds. Silicone is also sometimes used in breast implants, contact lenses, explosives and pyrotechnics.[93] Silly Putty was originally made by adding boric acid to silicone oil.[94] Other silicon compounds function as high-technology abrasives and new high-strength ceramics based upon silicon carbide. Silicon is a component of some superalloys.
Alloys[edit]
Elemental silicon is added to molten cast iron as ferrosilicon or silicocalcium alloys to improve performance in casting thin sections and to prevent the formation of cementite where exposed to outside air. The presence of elemental silicon in molten iron acts as a sink for oxygen, so that the steel carbon content, which must be kept within narrow limits for each type of steel, can be more closely controlled. Ferrosilicon production and use is a monitor of the steel industry, and although this form of elemental silicon is grossly impure, it accounts for 80% of the world’s use of free silicon. Silicon is an important constituent of electrical steel, modifying its resistivity and ferromagnetic properties.
The properties of silicon may be used to modify alloys with metals other than iron. «Metallurgical grade» silicon is silicon of 95–99% purity. About 55% of the world consumption of metallurgical purity silicon goes for production of aluminium-silicon alloys (silumin alloys) for aluminium part casts, mainly for use in the automotive industry. Silicon’s importance in aluminium casting is that a significantly high amount (12%) of silicon in aluminium forms a eutectic mixture which solidifies with very little thermal contraction. This greatly reduces tearing and cracks formed from stress as casting alloys cool to solidity. Silicon also significantly improves the hardness and thus wear-resistance of aluminium.[95][96]
Electronics[edit]
Silicon wafer with mirror finish
Most elemental silicon produced remains as a ferrosilicon alloy, and only approximately 20% is refined to metallurgical grade purity (a total of 1.3–1.5 million metric tons/year). An estimated 15% of the world production of metallurgical grade silicon is further refined to semiconductor purity.[96] This typically is the «nine-9» or 99.9999999% purity,[97] nearly defect-free single crystalline material.[98]
Monocrystalline silicon of such purity is usually produced by the Czochralski process, and is used to produce silicon wafers used in the semiconductor industry, in electronics, and in some high-cost and high-efficiency photovoltaic applications.[99] Pure silicon is an intrinsic semiconductor, which means that unlike metals, it conducts electron holes and electrons released from atoms by heat; silicon’s electrical conductivity increases with higher temperatures. Pure silicon has too low a conductivity (i.e., too high a resistivity) to be used as a circuit element in electronics. In practice, pure silicon is doped with small concentrations of certain other elements, which greatly increase its conductivity and adjust its electrical response by controlling the number and charge (positive or negative) of activated carriers. Such control is necessary for transistors, solar cells, semiconductor detectors, and other semiconductor devices used in the computer industry and other technical applications.[100] In silicon photonics, silicon may be used as a continuous wave Raman laser medium to produce coherent light.[101]
In common integrated circuits, a wafer of monocrystalline silicon serves as a mechanical support for the circuits, which are created by doping and insulated from each other by thin layers of silicon oxide, an insulator that is easily produced on Si surfaces by processes of thermal oxidation or local oxidation (LOCOS), which involve exposing the element to oxygen under the proper conditions that can be predicted by the Deal–Grove model. Silicon has become the most popular material for both high power semiconductors and integrated circuits because it can withstand the highest temperatures and greatest electrical activity without suffering avalanche breakdown (an electron avalanche is created when heat produces free electrons and holes, which in turn pass more current, which produces more heat). In addition, the insulating oxide of silicon is not soluble in water, which gives it an advantage over germanium (an element with similar properties which can also be used in semiconductor devices) in certain fabrication techniques.[102]
Monocrystalline silicon is expensive to produce, and is usually justified only in production of integrated circuits, where tiny crystal imperfections can interfere with tiny circuit paths. For other uses, other types of pure silicon may be employed. These include hydrogenated amorphous silicon and upgraded metallurgical-grade silicon (UMG-Si) used in the production of low-cost, large-area electronics in applications such as liquid crystal displays and of large-area, low-cost, thin-film solar cells. Such semiconductor grades of silicon are either slightly less pure or polycrystalline rather than monocrystalline, and are produced in comparable quantities as the monocrystalline silicon: 75,000 to 150,000 metric tons per year. The market for the lesser grade is growing more quickly than for monocrystalline silicon. By 2013, polycrystalline silicon production, used mostly in solar cells, was projected to reach 200,000 metric tons per year, while monocrystalline semiconductor grade silicon was expected to remain less than 50,000 tons per year.[96]
Quantum dots[edit]
Silicon quantum dots are created through the thermal processing of hydrogen silsesquioxane into nanocrystals ranging from a few nanometers to a few microns, displaying size dependent luminescent properties.[103][104] The nanocrystals display large Stokes shifts converting photons in the ultra-violet range to photons in the visible or infrared, depending on the particle size, allowing for applications in quantum dot displays and luminescent solar concentrators due to their limited self absorption. A benefit of using silicon based quantum dots over cadmium or indium is the non-toxic, metal-free nature of silicon.[105][106]
Another application of silicon quantum dots is for sensing of hazardous materials. The sensors take advantage of the luminescent properties of the quantum dots through quenching of the photoluminescence in the presence of the hazardous substance.[107] There are many methods used for hazardous chemical sensing with a few being electron transfer, fluorescence resonance energy transfer, and photocurrent generation.[108] Electron transfer quenching occurs when the lowest unoccupied molecular orbital (LUMO) is slightly lower in energy than the conduction band of the quantum dot, allowing for the transfer electrons between the two, preventing recombination of the holes and electrons within the nanocrystals. The effect can also be achieved in reverse with a donor molecule having its highest occupied molecular orbital (HOMO) slightly higher than a valence band edge of the quantum dot, allowing electrons to transfer between them, filling the holes and preventing recombination. Fluorescence resonance energy transfer occurs when a complex forms between the quantum dot and a quencher molecule. The complex will continue to absorb light but when the energy is converted to the ground state it does not release a photon, quenching the material. The third method uses different approach by measuring the photocurrent emitted by the quantum dots instead of monitoring the photoluminescent display. If the concentration of the desired chemical increases then the photocurrent given off by the nanocrystals will change in response.[109]
Biological role[edit]
A diatom, enclosed in a silica cell wall
Although silicon is readily available in the form of silicates, very few organisms use it directly. Diatoms, radiolaria, and siliceous sponges use biogenic silica as a structural material for their skeletons. Some plants accumulate silica in their tissues and require silicon for their growth, for example rice. Silicon may be taken up by plants as orthosilicic acid (also known as monosilicic acid) and transported through the xylem, where it forms amorphous complexes with components of the cell wall. This has been shown to improve cell wall strength and structural integrity in some plants, thereby reducing insect herbivory and pathogenic infections. In certain plants, silicon may also upregulate the production of volatile organic compounds and phytohormones which play a significant role in plant defense mechanisms.[110][111][112] In more advanced plants, the silica phytoliths (opal phytoliths) are rigid microscopic bodies occurring in the cell.[113][114][111]
Several horticultural crops are known to protect themselves against fungal plant pathogens with silica, to such a degree that fungicide application may fail unless accompanied by sufficient silicon nutrition. Silicaceous plant defense molecules activate some phytoalexins, meaning some of them are signalling substances producing acquired immunity. When deprived, some plants will substitute with increased production of other defensive substances.[111]
Life on Earth is largely composed of carbon, but astrobiology considers that extraterrestrial life may have other hypothetical types of biochemistry. Silicon is considered an alternative to carbon, as it can create complex and stable molecules with four covalent bonds, required for a DNA-analog, and it is available in large quantities.[115]
Marine microbial influences[edit]
Diatoms uses silicon in the biogenic silica (BSIO
2) form,[116] which is taken up by the silicon transport protein (SIT) to be predominantly used in the cell wall structure as frustules.[117] Silicon enters the ocean in a dissolved form such as silicic acid or silicate.[118] Since diatoms are one of the main users of these forms of silicon, they contribute greatly to the concentration of silicon throughout the ocean. Silicon forms a nutrient-like profile in the ocean due to the diatom productivity in shallow depths.[118] Therefore, less concentration of silicon in the upper ocean and more concentrations of silicon in the deep/lower ocean.
Diatom productivity in the upper ocean contribute to the amount of silicon exported to the lower ocean.[119] When diatom cells are lysed in the upper ocean, their nutrients such as iron, zinc, and silicon, are brought to the lower ocean through a process called marine snow. Marine snow involves the downward transfer of particulate organic matter by vertical mixing of dissolved organic matter.[120] It has been suggested that silicon is considered crucial to diatom productivity and as long as there is silicic acid available for diatoms to use, the diatoms can contribute to other important nutrient concentrations in the deep ocean as well.[121]
In coastal zones, diatoms serve as the major phytoplanktonic organisms and greatly contribute to biogenic silica production. In the open ocean, however, diatoms have a reduced role in global annual silica production. Diatoms in North Atlantic and North Pacific subtropical gyres only contribute about 5-7% of global annual marine silica production. The Southern Ocean produces about one-third of global marine biogenic silica.[87] The Southern Ocean is referred to as having a «biogeochemical divide»[122] since only minuscule amounts of silicon are transported out of this region.
Human nutrition[edit]
There is some evidence that silicon is important to human health for their nail, hair, bone, and skin tissues,[123] for example, in studies that demonstrate that premenopausal women with higher dietary silicon intake have higher bone density, and that silicon supplementation can increase bone volume and density in patients with osteoporosis.[124] Silicon is needed for synthesis of elastin and collagen, of which the aorta contains the greatest quantity in the human body,[125] and has been considered an essential element;[126] nevertheless, it is difficult to prove its essentiality, because silicon is very common, and hence, deficiency symptoms are difficult to reproduce.[127][128]
Silicon is currently under consideration for elevation to the status of a «plant beneficial substance by the Association of American Plant Food Control Officials (AAPFCO).»[129][130]
Safety[edit]
People may be exposed to elemental silicon in the workplace by breathing it in, swallowing it, or having contact with the skin or eye. In the latter two cases, silicon poses a slight hazard as an irritant. It is hazardous if inhaled.[131] The Occupational Safety and Health Administration (OSHA) has set the legal limit for silicon exposure in the workplace as 15 mg/m3 total exposure and 5 mg/m3 respiratory exposure over an eight-hour workday. The National Institute for Occupational Safety and Health (NIOSH) has set a recommended exposure limit (REL) of 10 mg/m3 total exposure and 5 mg/m3 respiratory exposure over an eight-hour workday.[132] Inhalation of crystalline silica dust may lead to silicosis, an occupational lung disease marked by inflammation and scarring in the form of nodular lesions in the upper lobes of the lungs.[133]
See also[edit]
- Amorphous silicon
- Black silicon
- Covalent superconductors
- List of countries by silicon production
- List of silicon producers
- Monocrystalline silicon
- Polycrystalline silicon
- Printed silicon electronics
- Silicene
- Silicon nanowire
- Silicon tombac
- Silicon Valley
- Transistor
References[edit]
- ^ «Standard Atomic Weights: Silicon». CIAAW. 2009.
- ^ «New Type of Zero-Valent Tin Compound». Chemistry Europe. 27 August 2016.
- ^ Ram, R. S.; et al. (1998). «Fourier Transform Emission Spectroscopy of the A2D–X2P Transition of SiH and SiD» (PDF). J. Mol. Spectr. 190 (2): 341–352. doi:10.1006/jmsp.1998.7582. PMID 9668026.
- ^ Eranna, Golla (2014). Crystal Growth and Evaluation of Silicon for VLSI and ULSI. CRC Press. p. 7. ISBN 978-1-4822-3281-3.
- ^ Magnetic susceptibility of the elements and inorganic compounds, in Lide, D. R., ed. (2005). CRC Handbook of Chemistry and Physics (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- ^ Weast, Robert (1984). CRC, Handbook of Chemistry and Physics. Boca Raton, Florida: Chemical Rubber Company Publishing. pp. E110. ISBN 0-8493-0464-4.
- ^ a b c d Hopcroft, Matthew A.; Nix, William D.; Kenny, Thomas W. (2010). «What is the Young’s Modulus of Silicon?». Journal of Microelectromechanical Systems. 19 (2): 229. doi:10.1109/JMEMS.2009.2039697.
- ^ Weeks, Mary Elvira (1932). «The discovery of the elements: XII. Other elements isolated with the aid of potassium and sodium: beryllium, boron, silicon, and aluminum». Journal of Chemical Education. 9 (8): 1386–1412. Bibcode:1932JChEd…9.1386W. doi:10.1021/ed009p1386.
- ^ Voronkov, M. G. (2007). «Silicon era». Russian Journal of Applied Chemistry. 80 (12): 2190. doi:10.1134/S1070427207120397.
- ^ Kamal 2022
- ^ Cutter, Elizabeth G. (1978). Plant Anatomy. Part 1 Cells and Tissues (2nd ed.). London: Edward Arnold. ISBN 978-0-7131-2639-6.
- ^ «Silicon». Encyclopedia Britannica. Retrieved 22 August 2019.
- ^ In his table of the elements, Lavoisier listed five «salifiable earths» (i.e., ores that could be made to react with acids to produce salts (salis = salt, in Latin): chaux (calcium oxide), magnésie (magnesia, magnesium oxide), baryte (barium sulfate), alumine (alumina, aluminium oxide), and silice (silica, silicon dioxide). About these «elements», Lavoisier speculates: «We are probably only acquainted as yet with a part of the metallic substances existing in nature, as all those which have a stronger affinity to oxygen than carbon possesses, are incapable, hitherto, of being reduced to a metallic state, and consequently, being only presented to our observation under the form of oxyds, are confounded with earths. It is extremely probable that barytes, which we have just now arranged with earths, is in this situation; for in many experiments it exhibits properties nearly approaching to those of metallic bodies. It is even possible that all the substances we call earths may be only metallic oxyds, irreducible by any hitherto known process.» – from Lavoisier (1799). Elements of Chemistry. Translated by Robert Kerr (4 ed.). Edinburgh, Scotland: William Creec. p. 218. (The original passage appears in: Lavoisier (1789). Traité Élémentaire de Chimie. Vol. 1. Paris, France: Cuchet. p. 174.
- ^ a b c d e Greenwood & Earnshaw 1997, p. 328.
- ^ Davy, Humphry (1808). «Electro chemical researches, on the decomposition of the earths; with observations on the metals obtained from the alkaline earths, and on the amalgam procured from ammonia». Philosophical Transactions of the Royal Society of London. W. Bowyer and J. Nichols. 98: 333–370.. On p. 353 Davy coins the name «silicium» : «Had I been so fortunate as to have obtained more certain evidences on this subject, and to have procured the metallic substances I was in search of, I should have proposed for them the names of silicium [silicon], alumium [aluminium], zirconium, and glucium [beryllium].»
- ^ «14 Silicon». Elements.vanderkrogt.net. Retrieved 2008-09-12.
- ^ Gay-Lussac, Joseph Louis; Thénard, Louis Jacques baron (1811). Recherches physico-chimiques, faites sur la pile: sur la préparation chimique et les propriétés du potassium et du sodium; sur la décomposition de l’acide boracique; sur les acides fluorique, muriatique et muriatique oxigéné; sur l’action chimique de la lumière; sur l’analyse végétale et animale, etc (in French). Deterville. pp. 313–314; vol. 2, pp. 55–65.
- ^ Thomson, Thomas; Baldwin, Charles; Blackwood, William; Baldwin, Cradock; Bell & Bradfute, bookseller; Hodges & McArthur, bookseller (1817). A system of chemistry : in four volumes. University of Wisconsin — Madison. London : Printed for Baldwin, Craddock, and Joy, Paternoster-Row; William Blackwood, and Bell and Bradfute, Edinburgh; and Hodges and Macarthur, Dublin. p. 252.: «The base of silica has been usually considered as a metal, and called silicium. But as there is not the smallest evidence for its metallic nature, and as it bears a close resemblance to boron and carbon, it is better to class it along with these bodies, and to give it the name of silicon.»
- ^ See
- Berzelius announced his discovery of silicon («silicium») in: Berzelius, J. (presented: 1823; published: 1824) «Undersökning af flusspatssyran och dess märkvärdigaste föreningar» (Investigation of hydrofluoric acid and of its most noteworthy compounds), Kongliga Vetenskaps-Academiens Handlingar [Proceedings of the Royal Science Academy], 12 : 46–98. The isolation of silicon and its characterization are detailed in the section titled «Flussspatssyrad kisseljords sönderdelning med kalium,» pp. 46–68.
- The above article was printed in German in: J.J. Berzelius (1824) «II. Untersuchungen über Flussspathsäure und deren merkwürdigsten Verbindungen» (II. Investigations of hydrofluoric acid and its most noteworthy compounds), Annalen der Physik, 77: 169–230. The isolation of silicon is detailed in the section titled: «Zersetzung der flussspaths. Kieselerde durch Kalium» (Decomposition of silicate fluoride by potassium), pp. 204–210.
- The above article was reprinted in French in: Berzelius (1824) «Décomposition du fluate de silice par le potassium» (Decomposition of silica fluoride by potassium), Annales de Chimie et de Physique, 27: 337–359.
- Reprinted in English in: «On the mode of obtaining silicium, and on the characters and properties of that substance». The Philosophical Magazine and Journal: Comprehending Various Branches of Science, the Liberal and Fine Arts, Agriculture, Manufactures, and Commerce. Richard Taylor and Company. 65: 254–267. 1825.
- ^ Weeks, Mary Elvira (1932). «The discovery of the elements: XII. Other elements isolated with the aid of potassium and sodium: beryllium, boron, silicon, and aluminum». Journal of Chemical Education. 9 (8): 1386–1412. Bibcode:1932JChEd…9.1386W. doi:10.1021/ed009p1386.
- ^ Voronkov, M.G. (2007). «Silicon era». Russian Journal of Applied Chemistry. 80 (12): 2190. doi:10.1134/S1070427207120397. S2CID 195240638.
- ^ a b Kipping, Frederic Stanley (1937-03-01). «The bakerian lecture organic derivatives of silicon». Proceedings of the Royal Society of London. Series A — Mathematical and Physical Sciences. 159 (896): 139–148. Bibcode:1937RSPSA.159..139K. doi:10.1098/rspa.1937.0063.
- ^ Muller, Richard (January 1965). «One hundred years of organosilicon chemistry». Journal of Chemical Education. 42 (1): 41. Bibcode:1965JChEd..42…41M. doi:10.1021/ed042p41. ISSN 0021-9584.
- ^ In 1854, Deville was trying to prepare aluminium metal from aluminium chloride that was heavily contaminated with silicon chloride. Deville used two methods to prepare aluminium: heating aluminium chloride with sodium metal in an inert atmosphere (of hydrogen); and melting aluminum chloride with sodium chloride and then electrolyzing the mixture. In both cases, pure silicon was produced: the silicon dissolved in the molten aluminium, but crystallized upon cooling. Dissolving the crude aluminum in hydrochloric acid revealed flakes of crystallized silicon. See: Henri Sainte-Claire Deville (1854) «Note sur deux procédés de préparation de l’aluminium et sur une nouvelle forme du silicium» (Note on two procedures for the preparation of aluminium and on a new form of silicon), Comptes rendus, 39: 321–326.
Subsequently, Deville obtained crystalline silicon by heating the chloride or fluoride of silicon with sodium metal, isolating the amorphous silicon, then melting the amorphous form with salt and heating the mixture until most of the salt evaporated. See: Sainte-Claire Deville, H. (1855). «Du silicium et du titane (On silicon and titanium)». Comptes rendus. 40: 1034–1036. - ^ «Information on silicon – history, thermodynamic, chemical, physical and electronic properties». Etacude. Retrieved 2021-06-08.
- ^ «Silicon: History». Nautilus.fis.uc.pt. 2011-07-27. Archived from the original on July 27, 2011.
- ^ a b Aufray, B.; Kara, A.; Vizzini, S. B.; Oughaddou, H.; LéAndri, C.; Ealet, B.; Le Lay, G. (2010). «Graphene-like silicon nanoribbons on Ag(110): A possible formation of silicene». Applied Physics Letters. 96 (18): 183102. Bibcode:2010ApPhL..96r3102A. doi:10.1063/1.3419932.
- ^ a b Lalmi, B.; Oughaddou, H.; Enriquez, H.; Kara, A.; Vizzini, S. B.; Ealet, B. N.; Aufray, B. (2010). «Epitaxial growth of a silicene sheet». Applied Physics Letters. 97 (22): 223109. arXiv:1204.0523. Bibcode:2010ApPhL..97v3109L. doi:10.1063/1.3524215. S2CID 118490651.
- ^ a b c d e f g h i j k l m n Greenwood & Earnshaw 1997, p. 330.
- ^ Greenwood & Earnshaw 1997, pp. 337–340
- ^ a b «Timeline». The Silicon Engine. Computer History Museum. Retrieved 22 August 2019.
- ^ a b «1901: Semiconductor Rectifiers Patented as «Cat’s Whisker» Detectors». The Silicon Engine. Computer History Museum. Retrieved 23 August 2019.
- ^ «1947: Invention of the Point-Contact Transistor». The Silicon Engine. Computer History Museum. Retrieved 23 August 2019.
- ^ «1954: Morris Tanenbaum fabricates the first silicon transistor at Bell Labs». The Silicon Engine. Computer History Museum. Retrieved 23 August 2019.
- ^ Bassett, Ross Knox (2007). To the Digital Age: Research Labs, Start-up Companies, and the Rise of MOS Technology. Johns Hopkins University Press. pp. 22–23. ISBN 978-0-8018-8639-3.
- ^ Saxena, A. (2009). Invention of integrated circuits: untold important facts. International series on advances in solid state electronics and technology. World Scientific. pp. 96–97. ISBN 978-981-281-445-6.
- ^ a b Feldman, Leonard C. (2001). «Introduction». Fundamental Aspects of Silicon Oxidation. Springer Science & Business Media. pp. 1–11. ISBN 978-3-540-41682-1.
- ^ Dabrowski, Jarek; Müssig, Hans-Joachim (2000). «1.2. The Silicon Age». Silicon Surfaces and Formation of Interfaces: Basic Science in the Industrial World. World Scientific. pp. 3–13. ISBN 978-981-02-3286-3.
- ^ Siffert, Paul; Krimmel, Eberhard (2013). «Preface». Silicon: Evolution and Future of a Technology. Springer Science & Business Media. ISBN 978-3-662-09897-4.
- ^ Uskali, T.; Nordfors, D. (23 May 2007). «The role of journalism in creating the metaphor of Silicon Valley» (PDF). Innovation Journalism 4 Conference, Stanford University. Archived from the original (PDF) on 2012-09-07. Retrieved 2016-08-08.
- ^ «Silicon and Germanium». hyperphysics.phy-astr.gsu.edu. Retrieved 2021-06-07.
- ^ King 1995, pp. xiii–xviii
- ^ Greenwood & Earnshaw 1997, pp. 372.
- ^ a b c d e f g h i j Greenwood & Earnshaw 1997, p. 331.
- ^ Vladimir E. Dmitrienko and Viacheslav A. Chizhikov (2020). «An infinite family of bc8-like metastable phases in silicon». Phys. Rev. B. 101 (24): 245203. arXiv:1912.10672. Bibcode:2020PhRvB.101x5203D. doi:10.1103/PhysRevB.101.245203. S2CID 209444444.
- ^ a b c d e f g Audi, G.; Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S. (2017). «The NUBASE2016 evaluation of nuclear properties» (PDF). Chinese Physics C. 41 (3): 030001. Bibcode:2017ChPhC..41c0001A. doi:10.1088/1674-1137/41/3/030001.
- ^ Jerschow, Alexej. «Interactive NMR Frequency Map». New York University. Retrieved 2011-10-20.
- ^ Seitenzahl, Ivo Rolf; Townsley, Dean M. (2017). Nucleosynthesis in Thermonuclear Supernovae. Handbook of Supernovae. pp. 1955–1978. arXiv:1704.00415. Bibcode:2017hsn..book.1955S. doi:10.1007/978-3-319-21846-5_87. ISBN 978-3-319-21845-8. S2CID 118993185.
- ^ Khokhlov, A. M.; Oran, E. S.; Wheeler, J. C. (April 1997). «Deflagration‐to‐Detonation Transition in Thermonuclear Supernovae». The Astrophysical Journal. 478 (2): 678–688. arXiv:astro-ph/9612226. Bibcode:1997ApJ…478..678K. doi:10.1086/303815. S2CID 53486905.
- ^ Cameron, A.G.W. (1973). «Abundance of the Elements in the Solar System» (PDF). Space Science Reviews. 15 (1): 121–146. Bibcode:1973SSRv…15..121C. doi:10.1007/BF00172440. S2CID 120201972. Archived from the original (PDF) on 2011-10-21.
- ^ Reynolds, B. C. (June 2009). «Modeling the modern marine δ 30 Si distribution: MODELING THE MODERN MARINE δ 30 Si DISTRIBUTION». Global Biogeochemical Cycles. 23 (2): 1–13. doi:10.1029/2008GB003266. S2CID 128652214.
- ^ Stapf, André; Gondek, Christoph; Kroke, Edwin; Roewer, Gerhard (2019), Yang, Deren (ed.), «Wafer Cleaning, Etching, and Texturization», Handbook of Photovoltaic Silicon, Berlin, Heidelberg: Springer Berlin Heidelberg, pp. 311–358, doi:10.1007/978-3-662-56472-1_17, ISBN 978-3-662-56471-4, S2CID 226945433, retrieved 2021-03-07
- ^ a b Grabmaier, J. (1982). Silicon Chemical Etching. Berlin, Heidelberg: Springer Berlin Heidelberg. ISBN 978-3-642-68765-5. OCLC 840294227.
- ^ Greenwood & Earnshaw 1997, pp. 331–5
- ^ Kaupp, Martin (1 December 2006). «The role of radial nodes of atomic orbitals for chemical bonding and the periodic table» (PDF). Journal of Computational Chemistry. 28 (1): 320–325. doi:10.1002/jcc.20522. PMID 17143872. S2CID 12677737. Retrieved 14 October 2016.
- ^ a b c King 1995, pp. 43–44
- ^ a b Greenwood & Earnshaw 1997, pp. 374.
- ^ Kaupp, Martin (2007). «The role of radial nodes of atomic orbitals for chemical bonding and the periodic table». Journal of Computational Chemistry. 28 (1): 320–325. doi:10.1002/jcc.20522. ISSN 0192-8651. PMID 17143872.
- ^ Greenwood & Earnshaw 1997, pp. 327–328.
- ^ a b Greenwood & Earnshaw 1997, pp. 359–361.
- ^ Greenwood & Earnshaw 1997, p. 335-337.
- ^ King 1995, pp. 45–47
- ^ Wiber, E. (1977). «Alfred Stock and the Renaissance of Inorganic Chemistry» (PDF). Pure Appl. Chem. 49 (6): 691–700. doi:10.1351/pac197749060691. S2CID 53313463.
- ^ Mellor, J.W. (1947). A Comprehensive Treatise on Inorganic and Theoretical Chemistry. Vol. VI, [C(Part II), Si, Silicates]. Longman, Green and Co. pp. 223–7. OCLC 1044702591.
- ^ Porterfield, W.W. (2013) [1993]. «4.8 Bonding in Elements». Inorganic Chemistry: A Unified Approach (2nd ed.). Elsevier. p. 219. ISBN 978-0-323-13894-9.
- ^ Wiberg, N.; Wiberg, E.; Holleman, A.F. (2001). «2.2.3 Higher Saturated Silanes». Inorganic Chemistry. Academic Press. p. 844. ISBN 0-12-352651-5.
- ^ King 1995, p. 47
- ^ Miller, R.D.; Michl, J. (1989). «Polysilane high polymers». Chemical Reviews. 89 (6): 1359. doi:10.1021/cr00096a006.
- ^ a b c d Greenwood & Earnshaw 1997, pp. 340.
- ^ a b c King 1995, p. 48
- ^ Greenwood & Earnshaw 1997, p. 342-347.
- ^ a b c Greenwood & Earnshaw 1997, p. 342.
- ^ a b c d e Greenwood & Earnshaw 1997, p. 347.
- ^ Geological Survey (U.S.) (1975). Geological Survey professional paper.
- ^ Korzhinsky, M.A.; Tkachenko, S.I.; Shmulovich, K.I.; Steinberg, G.S. (1995). «Native AI and Si formation» (PDF). Nature. 375 (6532): 544. Bibcode:1995Natur.375..544K. doi:10.1038/375544a0. ISSN 0028-0836. S2CID 39954119.
- ^ Cordua, Courtesy of Dr Bill (1998-01-10), English: PDF file entitled: «Silicon, Silica, Silicates and Silicone» (PDF), archived from the original (PDF) on 2016-04-18, retrieved 2016-03-29
- ^ Greenwood & Earnshaw 1997, p. 347-359.
- ^ a b c d Greenwood & Earnshaw 1997, p. 359.
- ^ a b c Greenwood & Earnshaw 1997, p. 334.
- ^ Cheung, Rebecca (2006). Silicon carbide microelectromechanical systems for harsh environments. Imperial College Press. p. 3. ISBN 978-1-86094-624-0.
- ^ Morkoç, H.; Strite, S.; Gao, G.B.; Lin, M.E.; Sverdlov, B.; Burns, M. (1994). «Large-band-gap SiC, III–V nitride, and II–VI ZnSe-based semiconductor device technologies». Journal of Applied Physics. 76 (3): 1363. Bibcode:1994JAP….76.1363M. doi:10.1063/1.358463.
- ^ a b c d e Greenwood & Earnshaw 1997, p. 361.
- ^ a b Clayden, pp. 668–77
- ^ Greenwood & Earnshaw 1997, p. 366.
- ^ a b Greenwood & Earnshaw 1997, p. 329.
- ^ Greenwood & Earnshaw 1997, pp. 329–330
- ^ a b c Tréguer, Paul J.; De La Rocha, Christina L. (3 January 2013). «The World Ocean Silica Cycle». Annual Review of Marine Science. 5 (1): 477–501. doi:10.1146/annurev-marine-121211-172346. PMID 22809182.
- ^ a b Tegen, Ina; Kohfeld, Karen (2006). Atmospheric transport of silicon. Island Press. pp. 81–91. ISBN 1-59726-115-7.
- ^ «Silicon Commodities Report 2011» (PDF). USGS. Retrieved 2011-10-20.
- ^ Zulehner, Neuer & Rau, p. 574
- ^ Kamali, A.R. (2019). «Ultra-fast shock-wave combustion synthesis of nanostructured silicon from sand with excellent Li storage performance». Sustainable Energy & Fuels. 3 (6): 1396–1405. doi:10.1039/C9SE00046A. S2CID 139986478.
- ^ Greenwood & Earnshaw 1997, p. 356.
- ^ Koch, E.C.; Clement, D. (2007). «Special Materials in Pyrotechnics: VI. Silicon – An Old Fuel with New Perspectives». Propellants, Explosives, Pyrotechnics. 32 (3): 205. doi:10.1002/prep.200700021.
- ^ Walsh, Tim (2005). «Silly Putty». Timeless toys: classic toys and the playmakers who created them. Andrews McMeel Publishing. ISBN 978-0-7407-5571-2.
- ^ Apelian, D. (2009). «Aluminum Cast Alloys: Enabling Tools for Improved Performance» (PDF). Wheeling, Illinois: North American Die Casting Association. Archived from the original (PDF) on 2012-01-06.
- ^ a b c Corathers, Lisa A. 2009 Minerals Yearbook. USGS
- ^ «Semi» SemiSource 2006: A supplement to Semiconductor International. December 2005. Reference Section: How to Make a Chip. Adapted from Design News. Reed Electronics Group.
- ^ SemiSource 2006: A supplement to Semiconductor International. December 2005. Reference Section: How to Make a Chip. Adapted from Design News. Reed Electronics Group.
- ^ Zulehner, Neuer & Rau, p. 590
- ^ Zulehner, Neuer & Rau, p. 573
- ^ Dekker, R; Usechak, N; Först, M; Driessen, A (2008). «Ultrafast nonlinear all-optical processes in silicon-on-insulator waveguides». Journal of Physics D. 40 (14): R249–R271. Bibcode:2007JPhD…40..249D. doi:10.1088/0022-3727/40/14/r01. S2CID 123008652.
- ^ Semiconductors Without the Quantum Physics. Electropaedia
- ^ Clark, Rhett J.; Aghajamali, Maryam; Gonzalez, Christina M.; Hadidi, Lida; Islam, Muhammad Amirul; Javadi, Morteza; Mobarok, Md Hosnay; Purkait, Tapas K.; Robidillo, Christopher Jay T.; Sinelnikov, Regina; Thiessen, Alyxandra N. (2017-01-10). «From Hydrogen Silsesquioxane to Functionalized Silicon Nanocrystals». Chemistry of Materials. 29 (1): 80–89. doi:10.1021/acs.chemmater.6b02667. ISSN 0897-4756.
- ^ Hessel, Colin M.; Henderson, Eric J.; Veinot, Jonathan G. C. (2007). «Hydrogen Silsesquioxane: A Molecular Precursor for Nanocrystalline Si—SiO2 Composites and Freestanding Hydride-Surface-Terminated Silicon Nanoparticles». ChemInform. 38 (10). doi:10.1002/chin.200710014. ISSN 1522-2667.
- ^ Lim, Cheol Hong; Han, Jeong-Hee; Cho, Hae-Won; Kang, Mingu (2014). «Studies on the Toxicity and Distribution of Indium Compounds According to Particle Size in Sprague-Dawley Rats». Toxicological Research. 30 (1): 55–63. doi:10.5487/TR.2014.30.1.055. ISSN 1976-8257. PMC 4007045. PMID 24795801.
- ^ Zou, Hui; Wang, Tao; Yuan, Junzhao; Sun, Jian; Yuan, Yan; Gu, Jianhong; Liu, Xuezhong; Bian, Jianchun; Liu, Zongping (2020-03-15). «Cadmium-induced cytotoxicity in mouse liver cells is associated with the disruption of autophagic flux via inhibiting the fusion of autophagosomes and lysosomes». Toxicology Letters. 321: 32–43. doi:10.1016/j.toxlet.2019.12.019. ISSN 0378-4274. PMID 31862506. S2CID 209435190.
- ^ Nguyen, An; Gonzalez, Christina M; Sinelnikov, Regina; Newman, W; Sun, Sarah; Lockwood, Ross; Veinot, Jonathan G C; Meldrum, Al (2016-02-10). «Detection of nitroaromatics in the solid, solution, and vapor phases using silicon quantum dot sensors». Nanotechnology. 27 (10): 105501. Bibcode:2016Nanot..27j5501N. doi:10.1088/0957-4484/27/10/105501. ISSN 0957-4484. PMID 26863492. S2CID 24292648.
- ^ Gonzalez, Christina M.; Veinot, Jonathan G. C. (2016-06-02). «Silicon nanocrystals for the development of sensing platforms». Journal of Materials Chemistry C. 4 (22): 4836–4846. doi:10.1039/C6TC01159D. ISSN 2050-7534.
- ^ Yue, Zhao; Lisdat, Fred; Parak, Wolfgang J.; Hickey, Stephen G.; Tu, Liping; Sabir, Nadeem; Dorfs, Dirk; Bigall, Nadja C. (2013-04-24). «Quantum-Dot-Based Photoelectrochemical Sensors for Chemical and Biological Detection». ACS Applied Materials & Interfaces. 5 (8): 2800–2814. doi:10.1021/am3028662. ISSN 1944-8244. PMID 23547912.
- ^ Kim, Sang Gyu; Kim, Ki Woo; Park, Eun Woo; Choi, Doil (2002). «Silicon-Induced Cell Wall Fortification of Rice Leaves: A Possible Cellular Mechanism of Enhanced Host Resistance to Blast». Phytopathology. 92 (10): 1095–103. doi:10.1094/PHYTO.2002.92.10.1095. PMID 18944220.
- ^ a b c Epstein, Emanuel (1999). «SILICON». Annual Review of Plant Physiology and Plant Molecular Biology. 50: 641–664. doi:10.1146/annurev.arplant.50.1.641. PMID 15012222.
- ^ Leroy, Nicolas; de Tombeur, Felix; Walgraffe, Yseult; Cornelis, Jean-Thomas; Verheggen, Francois (23 October 2019). «Silicon and plant natural defenses against insect pests: impact on plant volatile organic compounds and cascade effects on multitrophic interactions». Plants. 8 (444): 444. doi:10.3390/plants8110444. PMC 6918431. PMID 31652861.
- ^ Rahman, Atta-ur- (2008). «Silicon». Studies in Natural Products Chemistry. Vol. 35. p. 856. ISBN 978-0-444-53181-0.
- ^ Exley, C. (1998). «Silicon in life:A bioinorganic solution to bioorganic essentiality». Journal of Inorganic Biochemistry. 69 (3): 139–144. doi:10.1016/S0162-0134(97)10010-1.
- ^ Aguilera Mochón, Juan Antonio (2016). La vida no terrestre [The non-terrestrial life] (in Spanish). RBA. pp. 43–45. ISBN 978-84-473-8665-9.
- ^ Bidle, Kay D.; Manganelli, Maura; Azam, Farooq (2002-12-06). «Regulation of Oceanic Silicon and Carbon Preservation by Temperature Control on Bacteria». Science. 298 (5600): 1980–1984. Bibcode:2002Sci…298.1980B. doi:10.1126/science.1076076. ISSN 0036-8075. PMID 12471255. S2CID 216994.
- ^ Durkin, Colleen A.; Koester, Julie A.; Bender, Sara J.; Armbrust, E. Virginia (2016). «The evolution of silicon transporters in diatoms». Journal of Phycology. 52 (5): 716–731. doi:10.1111/jpy.12441. ISSN 1529-8817. PMC 5129515. PMID 27335204.
- ^ a b Dugdale, R. C.; Wilkerson, F. P. (2001-12-30). «Sources and fates of silicon in the ocean: the role of diatoms in the climate and glacial cycles». Scientia Marina. 65 (S2): 141–152. doi:10.3989/scimar.2001.65s2141. ISSN 1886-8134.
- ^ Baines, Stephen B.; Twining, Benjamin S.; Brzezinski, Mark A.; Krause, Jeffrey W.; Vogt, Stefan; Assael, Dylan; McDaniel, Hannah (December 2012). «Significant silicon accumulation by marine picocyanobacteria». Nature Geoscience. 5 (12): 886–891. Bibcode:2012NatGe…5..886B. doi:10.1038/ngeo1641. ISSN 1752-0908.
- ^ Turner, Jefferson T. (January 2015). «Zooplankton fecal pellets, marine snow, phytodetritus and the ocean’s biological pump». Progress in Oceanography. 130: 205–248. Bibcode:2015PrOce.130..205T. doi:10.1016/j.pocean.2014.08.005. ISSN 0079-6611.
- ^ Yool, Andrew; Tyrrell, Toby (2003). «Role of diatoms in regulating the ocean’s silicon cycle». Global Biogeochemical Cycles. 17 (4): n/a. Bibcode:2003GBioC..17.1103Y. CiteSeerX 10.1.1.394.3912. doi:10.1029/2002GB002018. ISSN 1944-9224. S2CID 16849373.
- ^ Marinov, I.; Gnanadesikan, A.; Toggweiler, J. R.; Sarmiento, J. L. (June 2006). «The Southern Ocean biogeochemical divide». Nature. 441 (7096): 964–967. Bibcode:2006Natur.441..964M. doi:10.1038/nature04883. PMID 16791191. S2CID 4428683.
- ^ Martin, Keith R. (2013). «Chapter 14. Silicon: The Health Benefits of a Metalloid». In Astrid Sigel; Helmut Sigel; Roland K.O. Sigel (eds.). Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. Vol. 13. Springer. pp. 451–473. doi:10.1007/978-94-007-7500-8_14. ISBN 978-94-007-7499-5. PMID 24470100.
- ^ Jugdaohsingh, R. (Mar–Apr 2007). «Silicon and bone health». The Journal of Nutrition, Health and Aging. 11 (2): 99–110. PMC 2658806. PMID 17435952.
- ^ Loeper, J.; Fragny, M. (1978). The Physiological Role of the Silicon and its AntiAtheromatous Action. Biochemistry of Silicon and Related Problems. pp. 281–296. doi:10.1007/978-1-4613-4018-8_13. ISBN 978-1-4613-4020-1.
- ^ Nielsen, Forrest H. (1984). «Ultratrace Elements in Nutrition». Annual Review of Nutrition. 4: 21–41. doi:10.1146/annurev.nu.04.070184.000321. PMID 6087860.
- ^ Lippard, Stephen J.; Jeremy M. Berg (1994). Principles of Bioinorganic Chemistry. Mill Valley, CA: University Science Books. p. 411. ISBN 978-0-935702-72-9.
- ^ Muhammad Ansar Farooq; Karl-Josef Dietz (2015). «Silicon as Versatile Player in Plant and Human Biology: Overlooked and Poorly Understood Muhammad Ansar Farooq and Karl-J». Front. Plant Sci. 6 (994): 994. doi:10.3389/fpls.2015.00994. PMC 4641902. PMID 26617630.
- ^ «AAPFCO Board of Directors 2006 Mid-Year Meeting» (PDF). Association of American Plant Food Control Officials. Archived from the original (PDF) on 6 January 2012. Retrieved 2011-07-18.
A presentation was made for Excell Minerals to recognize Silicon as a recognized plant nutrient
- ^ Miranda, Stephen R.; Barker, Bruce (August 4, 2009). «Silicon: Summary of Extraction Methods». Harsco Minerals. Archived from the original on November 12, 2012. Retrieved 2011-07-18.
- ^ Science Lab.com. «Material Safety Data Sheet: Silicon MSDS». sciencelab.com. Archived from the original on 23 March 2018. Retrieved 11 March 2018.
- ^ «CDC – NIOSH Pocket Guide to Chemical Hazards – Silicon». www.cdc.gov. Retrieved 2015-11-21.
- ^ Jane A. Plant; Nick Voulvoulis; K. Vala Ragnarsdottir (2012). Pollutants, Human Health and the Environment: A Risk Based Approach. Applied Geochemistry. Vol. 26. John Wiley & Sons. p. 273. Bibcode:2011ApGC…26S.238P. doi:10.1016/j.apgeochem.2011.03.113. ISBN 978-0-470-74261-7. Retrieved 24 August 2012.
Bibliography[edit]
- Clayden, Jonathan; Greeves, Nick; Warren, Stuart (2012). Organic Chemistry (2nd ed.). Oxford University Press. ISBN 978-0-19-927029-3.
- Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 978-0-08-037941-8.
- King, R. Bruce (1995). Inorganic Chemistry of Main Group Elements. Wiley-VCH. ISBN 978-0-471-18602-1.
- Zulehner, Werner; Neuer, Bernd; Rau, Gerhard. «Silicon». Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a23_721.
- Kamal, Kamal Y. (2022). «The Silicon Age: Trends in Semiconductor Devices Industry» (PDF). Journal of Engineering Science and Technology Review. 15 (1): 110–5. doi:10.25103/jestr.151.14. S2CID 249074588.
External links[edit]
- «Silicon Video — The Periodic Table of Videos — University of Nottingham». www.periodicvideos.com. Retrieved 2021-06-08.
- «CDC — NIOSH Pocket Guide to Chemical Hazards — Silicon». www.cdc.gov. Retrieved 2021-06-08.
- «Physical properties of Silicon (Si)». www.ioffe.ru. Retrieved 2021-06-08.
- The Story of Solar-Grade Silicon. Asianometry. 30 November 2022.
(Silicium) Si, химический элемент IV гр. периодич. системы, ат. н. 14, ат. м. 28,0855. Состоит из трех стабильных изотопов 28Si (92,27%), 29Si (4,68%) и 30Si (3,05%). Поперечное сечение захвата тепловых нейтронов 1,3.10-29 м 2. Конфигурация внеш. электронной оболочки <3s23p2;> степень окисления +4 (наиб. устойчива), +3, +2 и +1; энергии ионизации при последоват. переходе от Si° к Si4+ соотв. 8,1517, 16,342, 33,46 и 45,13 эВ; сродство к электрону 1,22 эВ; электроотрицательность по Полингу 1,8; атомный радиус 0,133, ионный радиус Si4+ (в скобках указаны координац. числа) 0,040 нм (4), 0,054 нм (6), ковалентный — 0,1175 нм. К.-второй после кислорода по распространенности в земной коре элемент (27,6% по массе). В своб. состоянии в природе не встречается, находится преим. в виде SiO2 (см. Кремния диоксид) или силикатов. В виде SiO2 К. входят в состав растит. и животных организмов (напр., скелетные части).
Свойства. Компактный К.- в-во серебристо-серого цвета с металлич. блеском. Кристаллич. решетка устойчивой модификации кубич. гранецентрированная типа алмаза, а=0,54307 нм, пространств. группа Fd3m,z=4. При высоких давлениях существуют др. полиморфные модификации: при 20 ГПа-К. I с тетрагон. решеткой (а=0,4686 нм, с=0,2585 нм), выше 20 ГПа-К. II с кубич. (а=0,644 нм) и К. III с гексагон. (а=0,380 нм, с=0,628 нм). При кристаллизации из газовой фазы на пов-стях с т-рой ниже 600 °С образуется аморфный К. Для кристаллич. Si т. пл. 1415 °С (плавится с уменьшением объема на 9%), т. кип. 3249 °С; плотн. 2,33 г/см 3; C0p20,16 Дж/(моль . К); DH0 пл 49,9 кДж/моль, DH0 исп 445,2 кДж/моль; S298 18,9 Дж/(моль . К); давление пара 0,046 Па (1415 °С); температурный коэф. линейного расширения 3,72.10-6 К -1 (291-1273 К) и -.0,6.10-6 К -1 (84 К); теплопроводность 95,5 Вт/(м . К); р 2,4-107 Ом м (25 °С); т-ра Дебая 645 К; e 12; диамагнетик, магн. восприимчивость Ч 3,9.10-6. При обычных условиях К. хрупок, выше 800 °С становится пластичным. К. прозрачен для И К излучения при длинах волн l>1 мкм; коэф. преломления 3,565 (l=1,05 мкм), 3,443 (l=2,6 мкм), 3,45 (l= 2-10 мкм); отражат. способность 0,3 (l>1,5 мкм). К. — полупроводник; ширина запрещенной зоны 1,21 эВ при т-ре ок. 0 К и 1,09 эВ при 300 К; концентрация носителей тока в К. с собственной проводимостью 1,5-1016 м -3 (300 К); температурная зависимость подвижности электронов и дырок [м 2/(В . с)] определяется соотв. выражениями: mn=4,0.105 Т -2,6 (300[T[400 К) и m р = 2,5.104T-2,3 (150[T[400 К); при 300 К mn= 0,145 м 2/(В . с), mp=0,048 м 2/(В . с), коэф. диффузии электронов 3,5.10-3 м 2/с, дырок — 1,3.10-3 м 2/с. Электрофиз. св-ва К. зависят от природы и концентрации присутствующих примесей и структурных дефектов. Для получения монокристаллов К. с дырочной проводимостью используют легирующие добавки В, Al, Ga, In (акцепторные примеси), с электронной проводимостью — Р, As, Sb (донорные примеси). Примеси Аu, Сu, Fe, Mn, V и нек-рые др. существенно снижают время жизни носителей тока в монокристаллах К. Макс, р-римость примесей в К. наблюдается при 1200-1300 °С и м. б. грубо оценена по значению коэф. распределения между твердым К. и его расплавом. Акцепторные примеси в К. имеют большие значения коэф. диффузии, чем донорные. Ряд примесей (Li, Сu, Аu) диффундирует по междоузлиям кристаллич. решетки с очень высокими скоростями. Для определения содержания примесей в К. высокой чистоты используют прецизионные методы: спектральный и активационный анализ, метод ЭПР и др. Производят монокристаллы К. без дислокаций диаметром до 0,156 м. Осн. дефекты в таких монокристаллах К.-скопления собств. междоузельных атомов, вакансий и атомов остаточных примесей. Для определения природы и содержания структурных дефектов в К. применяют избират. травление (в осн. смесью к-т: HF, HNO3 и СН 3 СООН), рентгеновский и др. методы. Электрич. св-ва К. могут сильно изменяться при термич. обработке. Так, нагревание монокристаллов, содержащих кислород, до 400-500 °С приводит к увеличению электронной проводимости, а при послед. нагревании до 1 1000-1200 °С этот эффект пропадает. Обычно термич. обработка приводит к существ. снижению времени жизни носителей тока. Для предотвращения вредного действия термич. обработки используют предварит. обработку пов-сти монокристаллов К. спец. орг. реактивами, отжиг в хлорсодержащей атмосфере, грубую шлифовку, бомбардировку ионами и др. методы. При низких т-рах К. химически инертен, при нагр. его реакц. способность резко возрастает. Особенно активен расплавленный К. Координац. число атома К. 4, иногда 6 (напр., во фторосиликатах, содержащих анион [SiF6]2-). Соед., где К. формально двухвалентен, по-видимому, содержат связь SiЧSi и, как правило, полимерны. Благодаря образующейся на пов-сти защитной оксидной пленке К. устойчив на воздухе даже при повыш. т-рах. Окисляется О 2 выше 400°С до SiO2 (см. также Кремния оксид). Стоек к действию к-т, взаимод. только со смесью HNO3 и фтористоводородной к-ты. Хорошо реагирует с р-рами щелочей с выделением Н 2 и образованием силикатов. Взаимод. с F2 уже при комнатной т-ре, с остальными галогенами — при 300-500 °С с образованием галогенидов SiX4 или Sin,X2n+2 (см. Кремния иодиды, Кремния фториды, Кремния хлориды). С парами S при 600 °С дает дисульфид SiS2, к-рый выше 600 °С переходит в моносульфид SiS; аналогичные, хотя и менее прочные соед., образует с Те и Se. С Н 2 К. непосредственно не реагирует, поэтому силаныSinH2n+2 получают косвенным путем — разложением силицидов. Аморфный К. обладает способностью растворять значит. кол-ва разл. газов, прежде всего Н 2. При этом образуется твердый р-р (до 47 ат. % водорода), называемый l-Si:H, к-рый обладает полупроводниковыми св-вами. С азотом выше 1000 °С К. образует кремния нитридSi3N4, с фосфором — фосфид SiP, с мышьяком — арсениды SiAs2 и SiAs, с углеродом -кремния карбидSiC, с бором — термически и химически стойкие бориды SiB3, SiB6 и SiB12. С большинством металлов дает тугоплавкие высокотвердые силициды. Об орг. производных К. см. Кремнийорганические полимеры, Кремнийорганические соединения, Кремнийэлементоорганические соединения.
Получение. К. производят восстановлением расплава SiO2 углеродом в дуговых печах при 1800°С. Чистота техн. продукта после спец. кислотной обработки ок. 99,9%. Очень небольшие кол-ва К. получают электролизом р-ров Na2SiF6 или K2SiF6 в расплавах. Для получения К. высокой чистоты техн. продукт хлорируют до SiQ4 или SiHCl3. Эти хлориды подвергают глубокой очистке ректификацией, сорбцией, путем частичного гидролиза и спец. термич. обработок, а затем восстанавливают при 1200-1300 °С высокочистым Н 2 в установках из нержавеющей стали или непрозрачного кварцевого стекла. Восстанавливаемый К. осаждают на прутки из К. высокой чистоты. Др. пром. метод получения К. высокой чистоты основан на разложении ок. 1000 °С SiH4, предварительно очищенного ректификацией. SiH4 синтезируют взаимод. Mg2Si с соляной или уксусной к-той, диспропор-ционированием SiH(OC2H5)3 в присут. Na или р-цией LiAlH4 с SiQ4 в эфире. Перечисл. методами получают К. с суммарным содержанием остаточных примесей 10-7-10-8 % по массе. Монокристаллы К. выращивают по методу Чохральского или бестигельной зонной плавкой (см. Монокристаллов выращивание). В первом случае процесс проводят в кварцевых тиглях в вакууме или инертной атмосфере с применением нагревателей из особо чистого графита. Масса исходной загрузки 60-100 кг, диаметр получаемых монокристаллов до 0,15 м, длина до 1,5-2,0 м. Зонную плавку проводят в глубоком вакууме или атмосфере особо чистого Н 2; этим способом получают наиб. чистые монокристаллы. Диаметр монокристаллов до 0,125 м, длина до 1,5 м. Легируют монокристаллы непосредственно в процессе выращивания. Для получения однородных монокристаллов, легированных фосфором, их часто облучают медленными нейтронами [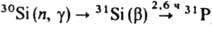 ]. Профилир. монокристаллы выращивают с помощью спец. формообразователя по способу Степанова, «горизонтальным сдергиванием» со своб. пов-сти расплава, кристаллизацией на спец. дендритных затравках. Поликристаллич. слитки получают направленной кристаллизацией в графитовой изложнице в условиях строго контролируемого тепло-отвода. Осн. пром. метод получения эпитаксиальных слоев и структур К.-хим. осаждение из газовой фазы с использованием смеси особо чистых SiCl4 и Н 2. Процесс проводят в проточных металлич. и кварцевых реакторах при 1250 °С и. атм. давлении с применением индукционного или радиационного нагрева. Эпитаксиальные слои наращивают на ориентированные и прошедшие спец. мех. и хим. обработку подложки из монокристаллич. К., размещаемые на кварцевом или графитовом (с покрытием SiC) пьедестале. Для снижения т-ры эпитаксиального наращивания в качестве источника К. используют SiH2Cl2, а сам процесс проводят при 6,6-9,3 кПа. Применяют также термич. разложение SiH4 (т-ра кристаллизации до 1000-1050 °C). Самую низкую т-ру кристаллизации (700-800 °С) обеспечивает метод мол. эпитаксии — наращивание из мол. пучков, получаемых нагреванием кремниевых заготовок электронным лучом в условиях глубокого вакуума (10-9-10-10 Па). Жидкофазную эпитаксию из р-ра К. в металлич. расплаве (наиб. часто Sn) проводят при 1100-1200°С. Пленки гидрогенизир. К., или a-Si:H, получают плазмохим. разложением SiH4, реактивным катодным распылением К. в атмосфере Н 2, а также хим. осаждением из газовой фазы с использованием смеси хлоридов К. и Н 2. Пленки наращивают на металлич. и стеклянные пластины при 200-400 °С.
]. Профилир. монокристаллы выращивают с помощью спец. формообразователя по способу Степанова, «горизонтальным сдергиванием» со своб. пов-сти расплава, кристаллизацией на спец. дендритных затравках. Поликристаллич. слитки получают направленной кристаллизацией в графитовой изложнице в условиях строго контролируемого тепло-отвода. Осн. пром. метод получения эпитаксиальных слоев и структур К.-хим. осаждение из газовой фазы с использованием смеси особо чистых SiCl4 и Н 2. Процесс проводят в проточных металлич. и кварцевых реакторах при 1250 °С и. атм. давлении с применением индукционного или радиационного нагрева. Эпитаксиальные слои наращивают на ориентированные и прошедшие спец. мех. и хим. обработку подложки из монокристаллич. К., размещаемые на кварцевом или графитовом (с покрытием SiC) пьедестале. Для снижения т-ры эпитаксиального наращивания в качестве источника К. используют SiH2Cl2, а сам процесс проводят при 6,6-9,3 кПа. Применяют также термич. разложение SiH4 (т-ра кристаллизации до 1000-1050 °C). Самую низкую т-ру кристаллизации (700-800 °С) обеспечивает метод мол. эпитаксии — наращивание из мол. пучков, получаемых нагреванием кремниевых заготовок электронным лучом в условиях глубокого вакуума (10-9-10-10 Па). Жидкофазную эпитаксию из р-ра К. в металлич. расплаве (наиб. часто Sn) проводят при 1100-1200°С. Пленки гидрогенизир. К., или a-Si:H, получают плазмохим. разложением SiH4, реактивным катодным распылением К. в атмосфере Н 2, а также хим. осаждением из газовой фазы с использованием смеси хлоридов К. и Н 2. Пленки наращивают на металлич. и стеклянные пластины при 200-400 °С.
Определение. Качественно К. обнаруживают по образованию (преим. в кислых средах) коллоидных р-ров гидратированного SiO2, окрашенных солей кремнемолибденовой к-ты H4[Si(Mo3O10)4]. Макроколичества К. (не менее 0,1% по массе) определяют гравиметрически, титриметрически и фотометрически. Гравиметрич. методы основаны на способности К. образовывать гель H2SiO3, к-рый затем высушивают и взвешивают. При титриметрич. определении К. переводят в H2SiF6, титруют щелочью или осаждают в виде малорастворимых солей H4[Si(Mo3O10)4] и определяют Мо в осадке. Большинство фотометрич. методов основано на переводе бесцв. H2SiO3 в желтую кремнемолибденовую к-ту, к-рую и определяют. Микроколичества К. определяют след. методами: эмиссионным спектральным (до 1-10-5 %), нейтронно-активационным (до 1.10-6 %), масс — спектрометрич. (до 1.10 -б %), атомно-абсорбционным с непламенной атомизацией (до 1.10-4 %). Применение. К.-один из осн. полупроводниковых материалов в электронике. Приборы на его основе могут работать при т-рах до 200 °С. Его используют для изготовления интегральных схем, диодов, транзисторов, солнечных батарей, фотоприемников, детекторов частиц в ядерной физике и др., а также линз в приборах ИК техники. В металлургии К. применяют как восстановитель (для получения силико-марганца, силикоалюминия и др.), при произ-ве ферросилиция, для раскисления — удаления растворенного в расплавленных металлах кислорода. К.-компонент электротехн. и др. сталей, чугунов, бронз, силуминов. К. и его соед. используют для получения кремнийорг. производных и силицидов ряда металлов. a-Si:H применяют для изготовления солнечных батарей, полевых транзисторов и др. Мировое произ-во К. (без СССР) для нужд полупроводникового приборостроения составляет ок. 5000 т/год поликристаллич. К. и ок. 2200 т/год монокристаллов (1984). К.-биогенный элемент. Он необходим для нормального роста и развития человека, животных, растений и микроорганизмов: является структурным элементом соединит. ткани, связывая макромолекулы мукополисахаридов и коллагена, играет существ. роль в метаболизме мн. растений и морских организмов, влияет на скорость минерализации и препятствует возникновению атеросклероза. Соед. К. токсичны. Вдыхание мельчайших частиц пыли SiO2 и др. соед. К. (напр., асбеста) вызывает опасную профессиональную болезнь — силикоз. К. получен впервые Ж. Л. Гей-Люссаком и Л. Ж. Тенаром в 1811. Лит.: Реньян В. Р.. Технология полупроводникового кремния, пер. с англ., М., 1969; Медведев С. А., Введение в технологию полупроводниковых материалов, М., 1970; Мильвидский М. Г., Полупроводниковые материалы в современной электронике, М., 1986; Нашельский А. Я.. Технология полупроводниковых материалов, М., 1987. М. Г. Мильвидский.
Химическая энциклопедия. — М.: Советская энциклопедия.
.
1988.
Кремний — химический элемент № (14). Он расположен в IVА группе третьем периоде Периодической системы.
Si14+14)2e)8e)4e
На внешнем слое атома кремния содержатся четыре валентных электрона. До его завершения не хватает четырёх электронов. Поэтому в соединениях с металлами кремнию характерна степень окисления (–4), а при взаимодействии с более электроотрицательными неметаллами он проявляет положительные степени окисления ( +2) или (+4).
По содержанию в земной коре кремний занимает второе место после кислорода. Земная кора более чем наполовину образована соединениями кремния. Распространены оксид кремния(IV)
SiO2
, силикаты и алюмосиликаты. Песок, кварц, горный хрусталь, аметист состоят из оксида. Гранит, полевой шпат, глина представляют собой силикаты и алюмосиликаты.
Входит кремний и в состав живых организмов. Его соединения придают прочность стеблям растений, содержатся в наружных покровах животных, образуют раковины и скелеты некоторых обитателей водной среды. У человека кремний присутствует в волосах и ногтях.

Рис. (1). Скелеты радиолярий
Кремний имеет атомную кристаллическую решётку, похожую на решётку алмаза. Каждый атом кремния в его кристаллах связан четырьмя ковалентными связями с соседними атомами. Благодаря такому строению у него высокая твёрдость.
Радиус атома кремния больше радиуса атома углерода, поэтому в его кристаллах электроны более свободны по сравнению с алмазом. Кремний проводит электрический ток, а его электропроводность увеличивается с повышением температуры или при освещении. Такие вещества относятся к полупроводникам.
В отличие от алмаза кремний представляет собой чёрно-серое непрозрачное вещество. У него высокая температура плавления ((1428) °С).

Рис. (2). Кремний
Получают кремний восстановлением его оксида коксом в электропечах:
В химических реакциях кремний может проявлять и окислительные, и восстановительные свойства. Окислительные свойства кремния выражены слабее, чем у остальных неметаллов.
- Взаимодействие с металлами.
При высокой температуре кремний реагирует с металлами с образованием силицидов:
В этой реакции кремний — окислитель.
- С водородом не реагирует.
С водородом кремний практически не реагирует по причине неустойчивости водородного соединения силана
SiH4
. Силан можно получить при гидролизе силицидов:
.
Он самовоспламеняется на воздухе и сгорает с образованием оксида кремния((IV)) и воды:
- Взаимодействие с кислородом.
Кремний горит в кислороде и проявляет в этой реакции восстановительные свойства:
- Взаимодействие с оксидами металлов.
Кремний способен восстанавливать некоторые металлы из их оксидов:
- Взаимодействие со щелочами.
В отличие от углерода кремний растворяется в концентрированных растворах щелочей c образованием силикатов и выделением водорода:
.
Применение кремния
-
используется в производстве полупроводников для электронной промышленности;
-
применяется для изготовления солнечных батарей;
-
входит в состав жаропрочных и кислотоустойчивых сплавов.

Рис. (3).Солнечные батареи
Источники:
Рис. 1. Скелеты радиолярий https://upload.wikimedia.org/wikipedia/commons/thumb/6/6e/Haeckel_Stephoidea.jpg/800px-Haeckel_Stephoidea.jpg
Рис. 2. Кремний https://upload.wikimedia.org/wikipedia/commons/e/e9/SiliconCroda.jpg
Рис. 3. Солнечные батареи https://cdn.pixabay.com/photo/2017/08/21/17/25/solar-photovoltaic-2666105_960_720.jpg
Толковый словарь русского языка. Поиск по слову, типу, синониму, антониму и описанию. Словарь ударений.
кремний
ТОЛКОВЫЙ СЛОВАРЬ
м.
Химический элемент, тёмно-серые кристаллы с металлическим блеском, являющийся составной частью большинства горных пород.
ТОЛКОВЫЙ СЛОВАРЬ УШАКОВА
КРЕ́МНИЙ, кремния, мн. нет, муж. (хим.). Химический элемент, входящий в состав большинства горных пород.
ТОЛКОВЫЙ СЛОВАРЬ ОЖЕГОВА
КРЕ́МНИЙ, -я, муж. Химический элемент, тёмно-серые кристаллы с металлическим блеском, одна из главных составных частей горных пород.
| прил. кремниевый, -ая, -ое.
СЛОВАРЬ СУЩЕСТВИТЕЛЬНЫХ
КРЕ́МНИЙ, -я, м
Химический элемент, темно-серые с металлическим блеском, кристаллы которого входят в состав большинства горных пород.
Кремний используется в производстве фотоэлементов, транзисторов как важнейший полупроводниковый материал. Кремний составляет 27,6 % массы земной коры.
ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
КРЕ́МНИЙ -я; м. [от греч. krēmnos — утёс, скала] Химический элемент (Si), тёмно-серые с металлическим блеском кристаллы которого входят в состав большинства горных пород.
◁ Кре́мниевый, -ая, -ое. К-ые соли. Кре́мни́стый (см. 2.К.; 1 зн.).
* * *
кре́мний (лат. Silicium), химический элемент IV группы периодической системы. Тёмно-серые кристаллы с металлическим блеском; плотность 2,33 г/см3, tпл 1415ºC. Стоек к химическим воздействиям. Составляет 27,6% массы земной коры (2-е место среди элементов), главные минералы — кремнезём и силикаты. Один из важнейших полупроводниковых материалов (транзисторы, термисторы, фотоэлементы). Составная часть многих сталей и других сплавов (повышает механическую прочность и устойчивость к коррозии, улучшает литейные свойства).

* * *
КРЕМНИЙ — КРЕ́МНИЙ (лат. Silicium от silex — кремень), Si (читается «силициум», но в настоящее время довольно часто и как «си»), химический элемент с атомным номером 14, атомная масса 28,0855. Русское название происходит от греческого kremnos — утес, гора.
Природный кремний состоит из смеси трех стабильных нуклидов (см. НУКЛИД) с массовыми числами 28 (преобладает в смеси, его в ней 92,27% по массе), 29 (4,68%) и 30 (3,05%). Конфигурация внешнего электронного слоя нейтрального невозбужденного атома кремния 3s2р2. В соединениях обычно проявляет степень окисления +4 (валентность IV) и очень редко +3, +2 и +1 (валентности соответственно III, II и I). В периодической системе Менделеева кремний расположен в группе IVA (в группе углерода), в третьем периоде.
Радиус нейтрального атома кремния 0,133 нм. Энергии последовательной ионизации атома кремния 8,1517, 16,342, 33,46 и 45,13 эВ, сродство к электрону 1,22 эВ. Радиус иона Si4+ при координационном числе 4 (наиболее распространенном в случае кремния) 0,040 нм, при координационном числе 6 — 0,054 нм. По шкале Полинга электроотрицательность кремния 1,9. Хотя кремний принято относить к неметаллам, он по ряду свойств занимает промежуточное положение между металлами и неметаллами.
В свободном виде — коричневый порошок или светло-серый компактный материал с металлическим блеском.
История открытия
Соединения кремния были известны человеку с незапамятных времен. Но с простым веществом кремнием человек познакомился всего около 200 лет тому назад. Фактически первыми исследователями, получившими кремний, были французы Ж. Л. Гей-Люссак (см. ГЕЙ-ЛЮССАК Жозеф Луи)и Л. Ж. Тенар (см. ТЕНАР Луи Жак) . Они в 1811 обнаружили, что нагревание фторида кремния с металлическим калием приводит к образованию буро-коричневого вещества:
SiF4+ 4K = Si + 4KF, однако сами исследователи правильного вывода о получении нового простого вещества не сделали. Честь открытия нового элемента принадлежит шведскому химику Й. Берцелиусу (см. БЕРЦЕЛИУС Йенс Якоб), который для получения кремния нагревал также с металлическим калием соединение состава K2SiF6. Он получил тот же аморфный порошок, что и французские химики, и в 1824 объявил о новом элементарном веществе, которое назвал «силиций». Кристаллический кремний был получен только в 1854 году французским химиком А. Э. Сент-Клер Девилем (см. СЕНТ-КЛЕР ДЕВИЛЬ Анри Этьен) .
Нахождение в природе
По распространенности в земной коре кремний среди всех элементов занимает второе место (после кислорода). На долю кремния приходится 27,7% массы земной коры. Кремний входит в состав нескольких сотен различных природных силикатов (см. СИЛИКАТЫ)и алюмосиликатов (см. АЛЮМОСИЛИКАТЫ). Широко распространен и кремнезем, или кремния диоксид (см. КРЕМНИЯ ДИОКСИД) SiO2 (речной песок (см. ПЕСОК) , кварц (см. КВАРЦ) , кремень (см. КРЕМЕНЬ) и др.), составляющий около 12% земной коры (по массе). В свободном виде кремний в природе не встречается.
Получение
В промышленности кремний получают, восстанавливая расплав SiO2 коксом при температуре около 1800°C в дуговых печах. Чистота полученного таким образом кремния составляет около 99,9%. Так как для практического использования нужен кремний более высокой чистоты, полученный кремний хлорируют. Образуются соединения состава SiCl4 и SiCl3H. Эти хлориды далее очищают различными способами от примесей и на заключительном этапе восстанавливают чистым водородом. Возможна также очистка кремния за счет предварительного получения силицида магния Mg2Si. Далее из силицида магния с помощью соляной или уксусной кислот получают летучий моносилан SiH4. Моносилан очищают далее ректификацией, сорбционными и др. методами, а затем разлагают на кремний и водород при температуре около 1000°C. Содержание примесей в получаемом этими методами кремнии снижается до 10-8-10-6% по массе.
Физические и химические свойства
Кристаллическая решетка кремния кубическая гранецентрированная типа алмаза, параметр а = 0,54307 нм (при высоких давлениях получены и другие полиморфные модификации кремния), но из-за большей длины связи между атомами Si-Si по сравнению с длиной связи С-С твердость кремния значительно меньше, чем алмаза.
Плотность кремния 2,33 кг/дм3. Температура плавления 1410°C, температура кипения 2355°C. Кремний хрупок, только при нагревании выше 800°C он становится пластичным веществом. Интересно, что кремний прозрачен к инфракрасному (ИК)-излучению.
Элементарный кремний — типичный полупроводник (см. ПОЛУПРОВОДНИКИ) . Ширина запрещенной зоны при комнатной температуре 1,09 эВ. Концентрация носителей тока в кремнии с собственной проводимостью при комнатной температуре 1,5·1016 м-3. На электрофизические свойства кристаллического кремния большое влияние оказывают содержащиеся в нем микропримеси. Для получения монокристаллов кремния с дырочной проводимостью в кремний вводят добавки элементов III-й группы — бора (см. БОР (химический элемент)), алюминия (см. АЛЮМИНИЙ), галлия (см. ГАЛЛИЙ) и индия (см. ИНДИЙ), с электронной проводимостью — добавки элементов V-й группы — фосфора (см. ФОСФОР), мышьяка (см. МЫШЬЯК) или сурьмы (см. СУРЬМА). Электрические свойства кремния можно варьировать, изменяя условия обработки монокристаллов, в частности, обрабатывая поверхность кремния различными химическими агентами.
Химически кремний малоактивен. При комнатной температуре реагирует только с газообразным фтором, при этом образуется летучий тетрафторид кремния SiF4. При нагревании до температуры 400-500°C кремний реагирует с кислородом с образованием диоксида SiO2, с хлором, бромом и иодом — с образованием соответствующих легко летучих тетрагалогенидов SiHal4.
С водородом кремний непосредственно не реагирует, соединения кремния с водородом — силаны (см. СИЛАНЫ) с общей формулой SinH2n+2 — получают косвенным путем. Моносилан SiH4 (его часто называют просто силаном) выделяется при взаимодействии силицидов металлов с растворами кислот, например:
Ca2Si + 4HCl = 2CaCl2 + SiH4
Образующийся в этой реакции силан SiH4 содержит примесь и других силанов, в частности, дисилана Si2H6 и трисилана Si3H8, в которых имеется цепочка из атомов кремния, связанных между собой одинарными связями (-Si-Si-Si-).
С азотом кремний при температуре около 1000°C образует нитрид Si3N4, с бором — термически и химически стойкие бориды SiB3, SiB6 и SiB12. Соединение кремния и его ближайшего аналога по таблице Менделеева — углерода — карбид кремния SiС (карборунд (см. КАРБОРУНД) ) характеризуется высокой твердостью и низкой химической активностью. Карборунд широко используется как абразивный материал.
При нагревании кремния с металлами возникают силициды (см. СИЛИЦИДЫ) . Силициды можно подразделить на две группы: ионно-ковалентные (силициды щелочных, щелочноземельных металлов и магния типа Ca2Si, Mg2Si и др.) и металлоподобные (силициды переходных металлов). Силициды активных металлов разлагаются под действием кислот, силициды переходных металлов химически стойки и под действием кислот не разлагаются. Металлоподобные силициды имеют высокие температуры плавления (до 2000°C). Наиболее часто образуются металлоподобные силициды составов MSi, M3Si2, M2Si3, M5Si3 и MSi2. Металлоподобные силициды химически инертны, устойчивы к действию кислорода даже при высоких температурах.
Диоксид кремния SiO2— кислотный оксид, не реагирующий с водой. Существует в виде нескольких полиморфных модификаций (кварц (см. КВАРЦ), тридимит, кристобалит, cтеклообразный SiO2). Из этих модификаций наибольшее практическое значение имеет кварц. Кварц обладает свойствами пьезоэлектрика (см. ПЬЕЗОЭЛЕКТРИЧЕСКИЕ МАТЕРИАЛЫ) , он прозрачен для ультрафиолетового (УФ) излучения. Характеризуется очень низким коэффициентом теплового расширения, поэтому изготовленная из кварца посуда не растрескивается при перепадах температуры до 1000 градусов.
Кварц химически стоек к действию кислот, но реагирует с плавиковой кислотой:
SiO2 + 6HF =H2[SiF6] + 2H2O
и газообразным фтороводородом HF:
SiO2 + 4HF =SiF4 + 2H2O
Эти две реакции широко используют для травления стекла.
При сплавлении SiO2 с щелочами и основными оксидами, а также с карбонатами активных металлов образуются силикаты (см. СИЛИКАТЫ) — соли не имеющих постоянного состава очень слабых нерастворимых в воде кремниевых кислот (см. КРЕМНИЕВЫЕ КИСЛОТЫ) общей формулы xH2O·ySiO2 (довольно часто в литературе не очень точно пишут не о кремниевых кислотах, а о кремниевой кислоте, хотя фактически речь при этом идет об одном и том же). Например, может быть получен ортосиликат натрия:
SiO2 + 4NaOH = (2Na2O)·SiO2 +2H2O,
метасиликат кальция:
SiO2 + СаО = СаО·SiO2
или смешанный силикат кальция и натрия:
Na2CO3 + CaCO3 + 6SiO2 = Na2O·CaO·6SiO2 + 2CO2
Из силиката Na2O·CaO·6SiO2 изготовляют оконное стекло.
Следует отметить, что большинство силикатов не имеет постоянного состава. Из всех силикатов растворимы в воде только силикаты натрия и калия. Растворы этих силикатов в воде называют растворимым стеклом. Из-за гидролиза эти растворы характеризуются сильно щелочной средой. Для гидролизованных силикатов характерно образование не истинных, а коллоидных растворов. При подкислении растворов силикатов натрия или калия выпадает студенистый белый осадок гидратированных кремниевых кислот.
Главным структурным элементом как твердого диоксида кремния, так и всех силикатов выступает группа [SiO4/2], в которой атом кремния Si окружен тетраэдром из четырех атомов кислорода О. При этом каждый атом кислорода соединен с двумя атомами кремния. Фрагменты [SiO4/2] могут быть связаны между собой по-разному. Среди силикатов по характеру связи в них фрагментов [SiO4/2] выделяют островные, цепочечные, ленточные, слоистые, каркасные и другие.
При восстановлении SiO2 кремнием при высоких температурах образуется монооксид кремния состава SiO.
Для кремния характерно образование кремнийорганических соединений (см. КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ) , в которых атомы кремния соединены в длинные цепочки за счет мостиковых атомов кислорода -О-, а к каждому атому кремния, кроме двух атомов О, присоединены еще два органических радикала R1 и R2 = CH3, C2H5, C6H5, CH2CH2CF3 и др.
Применение
Кремний используют как полупроводниковый материал. Кварц находит применение как пьезоэлектрик, как материал для изготовления жаропрочной химической (кварцевой) посуды, ламп УФ-излучения. Силикаты находят широкое применение как строительные материалы. Оконные стекла представляют собой аморфные силикаты. Кремнийорганические материалы характеризуются высокой износостойкостью и широко используются на практике в качестве силиконовых масел, клеев, каучуков, лаков.
Биологическая роль
Для некоторых организмов кремний является важным биогенным элементом (см. БИОГЕННЫЕ ЭЛЕМЕНТЫ). Он входит в состав опорных образований у растений и скелетных — у животных. В больших количествах кремний концентрируют морские организмы — диатомовые водоросли (см. ДИАТОМОВЫЕ ВОДОРОСЛИ), радиолярии (см. РАДИОЛЯРИИ), губки (см. ГУБКИ) . Мышечная ткань человека содержит (1-2)·10-2% кремния, костная ткань — 17·10-4%, кровь — 3,9 мг/л. С пищей в организм человека ежедневно поступает до 1 г кремния.
Соединения кремния не ядовиты. Но очень опасно вдыхание высокодисперсных частиц как силикатов, так и диоксида кремния, образующихся, например, при взрывных работах, при долблении пород в шахтах, при работе пескоструйных аппаратов и т. д. Микрочастицы SiO2, попавшие в легкие, в них кристаллизуются, а возникающие кристаллики разрушают легочную ткань и вызывают тяжелую болезнь — силикоз (см. СИЛИКОЗ). Чтобы не допустить попадания в легкие этой опасной пыли, следует использовать для защиты органов дыхания респиратор.
БОЛЬШОЙ ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
КРЕМНИЙ (лат. Silicium) — Si, химический элемент IV группы периодической системы, атомный номер 14, атомная масса 28,0855. Темно-серые кристаллы с металлическим блеском; плотность 2,33 г/см³, tпл 1415 .С. Стоек к химическим воздействиям. Составляет 27,6% массы земной коры (2-е место среди элементов), главные минералы — кремнезем и силикаты. Один из важнейших полупроводниковых материалов (транзисторы, термисторы, фотоэлементы). Составная часть многих сталей и других сплавов (повышает механическую прочность и устойчивость к коррозии, улучшает литейные свойства).
АКАДЕМИЧЕСКИЙ СЛОВАРЬ
-я, м.
Химический элемент, входящий в состав большинства горных пород.
[От греч. κρημνός — утес, скала]
ЭНЦИКЛОПЕДИЯ КОЛЬЕРА
Si (silicium)
химический элемент IVA подгруппы (C, Si, Ge, Sn и Pb) периодической системы элементов, неметалл. Кремний в свободном виде был выделен в 1811 Ж.Гей-Люссаком и Л.Тенаром при пропускании паров фторида кремния над металлическим калием, однако он не был описан ими как элемент. Шведский химик Й.Берцелиус в 1823 дал описание кремния, полученного им при обработке калиевой соли K2SiF6 металлическим калием при высокой температуре, однако лишь в 1854 кремний был получен в кристаллической форме А.Девилем. Кремний — второй по распространенности (после кислорода) элемент в земной коре, где он составляет более 25% (масс.). Встречается в природе в основном в виде песка, или кремнезема, который представляет собой диоксид кремния, и в виде силикатов (полевые шпаты M[[AlSi3O8]] (M = Na, K, Ba), каолинит Al4[[Si4O10]](OH)8, слюды). Кремний можно получить прокаливанием измельченного песка с алюминием или магнием; в последнем случае его отделяют от образующегося MgO растворением оксида магния в соляной кислоте. Технический кремний получают в больших количествах в электрических печах путем восстановления кремнезема углем или коксом. Полупроводниковый кремний получают восстановлением SiCl4 или SiHCl3 водородом с последующим разложением образующегося SiH4 при 400-600° С. Высокочистый кремний получают выращиванием монокристалла из расплава полупроводникового кремния по методу Чохральского или методом бестигельной зонной плавки кремниевых стержней
(см. также ЗОННАЯ ПЛАВКА).
Элементный кремний получают в основном для полупроводниковой техники, в остальных случаях он используется как легирующая добавка в производстве сталей и сплавов цветных металлов (например, для получения ферросилиция FeSi, который образуется при прокаливании смеси песка, кокса и оксида железа в электрической печи и применяется как раскислитель и легирующая добавка в производстве сталей и как восстановитель в производстве ферросплавов). Применение. Наибольшее применение кремний находит в производстве сплавов для придания прочности алюминию, меди и магнию и для получения ферросилицидов, имеющих важное значение в производстве сталей и полупроводниковой техники. Кристаллы кремния применяют в солнечных батареях и полупроводниковых устройствах — транзисторах и диодах. Кремний служит также сырьем для производства кремнийорганических соединений, или силоксанов, получаемых в виде масел, смазок, пластмасс и синтетических каучуков. Неорганические соединения кремния используют в технологии керамики и стекла, как изоляционный материал и пьезокристаллы.
См. также КРЕМНИЙОРГАНИЧЕСКИЕ ПОЛИМЕРЫ. СВОЙСТВА КРЕМНИЯ
Атомный номер 14 Атомная масса 28,086 Изотопы
стабильные 28, 29, 30
нестабильные 25, 26, 27, 31, 32, 33
Температура плавления, ° С 1410 Температура кипения, ° С 2355 Плотность, г/см3 2,33 Твердость (по Моосу) 7,0 Содержание в земной коре, % (масс.) 27,72 Степени окисления -4, +2, +4
Свойства. Кремний — темносерое, блестящее кристаллическое вещество, хрупкое и очень твердое, кристаллизуется в решетке алмаза. Это типичный полупроводник (проводит электричество лучше, чем изолятор типа каучука, и хуже проводника — меди). При высокой температуре кремний весьма реакционноспособен и взаимодействует с большинством элементов, образуя силициды, например силицид магния Mg2Si, и другие соединения, например SiO2 (диоксид кремния), SiF4 (тетрафторид кремния) и SiC (карбид кремния, карборунд). Кремний растворяется в горячем растворе щелочи с выделением водорода: Si + NaOH (r) Na4SiO4 + 2H2. 4 (тетрахлорид кремния) получают из SiO2 и CCl4 при высокой температуре; это бесцветная жидкость, кипящая при 58° С, легко гидролизуется, образуя хлороводородную (соляную) кислоту HCl и ортокремниевую кислоту H4SiO4 (это свойство используют для создания дымовых надписей: выделяющаяся HCl в присутствии аммиака образует белое облако хлорида аммония NH4Cl). Тетрафторид кремния SiF4 образуется при действии фтороводородной (плавиковой) кислоты на стекло: Na2SiO3 + 6HF -> 2NaF + SiF4 + 3H2O SiF4 гидролизуется, образуя ортокремниевую и гексафторокремниевую (H2SiF6) кислоты. H2SiF6 по силе близка к серной кислоте. Многие фторосиликаты металлов растворимы в воде (соли натрия, бария, калия, рубидия, цезия малорастворимы), поэтому HF используют для перевода минералов в раствор при выполнении анализов. Сама кислота H2SiF6 и ее соли ядовиты.
Диоксид кремния (кремниевый ангидрид). Природный диоксид кремния встречается преимущественно в форме кварца, хотя существуют и другие минералы — кристобалит, тридимит, китит, коусит. Кристаллический диоксид кремния широко распространен в природе в виде прозрачных бесцветных или окрашенных монокристаллов (горный хрусталь, аметист, дымчатый кварц, тридимит, кварцит, розовый кварц, агат, яшма, сердолик, кремень, опал и халцедон) и в форме обломочных пород (морской песок, гравий, галька, песчаник и конгломерат). Окраска аметиста объясняется примесями Mn и Fe, а дымчатого кварца — органическими включениями. Опал и кремень являются слабогидратированными формами SiO2. Аморфный кремнезем встречается в диатомовых отложениях на дне морей и океанов (трепел, кизельгур); эти отложения образовались из SiO2, входившего в состав диатомовых водорослей и некоторых инфузорий. Диатомитовая земля и трепел обнаружены в Калифорнии, Орегоне и в разных частях Европы. Ежегодно добывается до 2 млн. т SiO2 для производства абразивов, теплоизоляции, фильтрующих сред, наполнителя полимеров, красок и композиций. См. также КВАРЦ.
Кремниевые кислоты. Две оксокислоты кремния H4SiO4 (ортокремниевая) и H2SiO3 (метакремниевая, или кремниевая) существуют только в растворе и необратимо превращаются в SiO2, если выпарить воду. Другие кремниевые кислоты получаются за счет различного количества воды в их составе: H6Si2O7 (пирокремниевая кислота из двух молекул ортокремниевой кислоты), H2Si2O5 и H4Si3O8 (ди- и трикремниевая кислоты из двух и соответственно трех молекул метакремниевой кислоты). Все кислоты кремния слабые. При добавлении в раствор силиката серной кислоты образуется гель (желатинообразное вещество), при нагревании и высушивании которого остается твердый пористый продукт — силикагель, имеющий развитую поверхность и используемый как адсорбент газов, осушитель, катализатор и носитель катализаторов.
Силикаты (соли кремниевых кислот). В тетраэдрической структуре природных силикатов атом кремния окружен четырьмя атомами кислорода; ион щелочного или щелочноземельного металла, слишком малый по сравнению с кислородными атомами, размещается в пространстве между тетраэдрами. Иногда тетраэдры выстраиваются в протяженные цепи (например, асбест), иногда образуется слоистая структура (слюда), в других случаях формируется кольцевая структура (например, берилл). К природным силикатам относятся полевые шпаты, слюды, глины, асбест и др. Силикаты входят в состав горных пород: гранита, гнейса, базальта, различных сланцев и т.д. Многие драгоценные камни (изумруд, топаз, аквамарин и др.) — это прозрачные кристаллы силикатов. Силикаты в большинстве своем (кроме силикатов натрия и калия) нерастворимы в воде. Силикаты натрия и калия внешне напоминают стекло, поэтому их называют растворимым стеклом. Жидкое стекло — это водный раствор силиката натрия или калия. Силикат натрия получается сплавлением кварцевого песка со щелочью (NaOH) или содой (Na2CO3) или кипячением смеси кварца с NaOH под давлением. Коммерческий продукт содержит Na2SiO3 с непостоянной примесью SiO2. Растворимое стекло широко используется как наполнитель в мылах. Некоторые моющие средства тоже содержат силикат натрия. Жидкое стекло используют для придания влаго- и огнестойкости деревянным строениям, в технологии кислото- и огнеупорного цемента и бетона, керосинонепроницаемых штукатурок по бетону, для пропитывания тканей, для приготовления огнезащитных красок по дереву, для химического укрепления слабых грунтов.
Гидриды. Подобно углероду кремний образует ковалентные связи Si-Si и Si-H. Соединения, в которых атомы кремния соединены одинарной связью, называются силанами, а если атомы кремния соединены двойной связью, -силенами. Подобно углеводородам эти соединения образуют цепи и кольца. SiH4 называется моносилан, Si2H6 — дисилан, Si3H8 — трисилан, Si4H10 — тетрасилан и т.д. Соединения, в которых атомы кремния соединены через атом кислорода, называются силоксанами, а через атомы серы — силазанами. Силаны и силены могут образовывать связь с углеводородными радикалами и галогенами, например, метилдихлорсилан CH3SiHCl2. Все силаны могут самовозгораться, образуют взрывчатые смеси с воздухом и легко реагируют с водой.
См. также
КЕРАМИКА ПРОМЫШЛЕННАЯ;
ЭЛЕМЕНТЫ ХИМИЧЕСКИЕ;
СТЕКЛО;
ПОЛУПРОВОДНИКОВЫЕ ЭЛЕКТРОННЫЕ ПРИБОРЫ.
ЛИТЕРАТУРА
Андрианов К.А. Методы элементоорганической химии. Кремний. М., 1968 Воронков М.Г. и др. Кремний и жизнь. Рига, 1978 Самсонов Г.В. и др. Силициды. М., 1979 Айлер Р. Химия кремнезема. М., 1982
ИЛЛЮСТРИРОВАННЫЙ ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
КРЕМНИЙ (Silicium), Si, химический элемент IV группы периодической системы, атомный номер 14, атомная масса 28,0855; неметалл, tпл 1415°C. Кремний — второй после кислорода по распространенности на Земле элемент, содержание в земной коре 27,6% по массе. Один из основных полупроводниковых материалов в электронике, восстановитель при металлотермическом получении металлов, компонент сталей, чугунов и других сплавов. Открыт французскими учеными Ж. Гей-Люссаком и Л. Тенаром в 1811.
ОРФОГРАФИЧЕСКИЙ СЛОВАРЬ
СЛОВАРЬ УДАРЕНИЙ
ФОРМЫ СЛОВ
кре́мний, кре́мнии, кре́мния, кре́мниев, кре́мнию, кре́мниям, кре́мнием, кре́мниями, кре́мниях
СИНОНИМЫ
сущ., кол-во синонимов: 6
МОРФЕМНО-ОРФОГРАФИЧЕСКИЙ СЛОВАРЬ
ГРАММАТИЧЕСКИЙ СЛОВАРЬ
СКАНВОРДЫ
— В 2008 году международной группе учёных удалось получить силицен — слой этого элемента, толщиной в один атом.
— Химический элемент в составе жидкого стекла.
— Какой химический элемент присутствует в силиконовых имплантантах?
— Химический элемент, Si.
— Полупроводник.
— Элемент, который впервые выделен в чистом виде в 1811 году французскими учёными Жозефом Луи Гей-Люссаком и Луи Жаком Тенаром.
ПОЛЕЗНЫЕ СЕРВИСЫ
кремний-каучук
СЛИТНО. РАЗДЕЛЬНО. ЧЕРЕЗ ДЕФИС
кре/мний-каучу/к, кре/мний-каучу/ка
ПОЛЕЗНЫЕ СЕРВИСЫ
кремний-каучуковый
СЛИТНО. РАЗДЕЛЬНО. ЧЕРЕЗ ДЕФИС
ПОЛЕЗНЫЕ СЕРВИСЫ
кремнийорганические жидкости
ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
Кремнийоргани́ческие жи́дкости (силиконовые масла), один из видов кремнийорганических полимеров. Применяются в качестве гидравлических жидкостей, гидрофобизаторов, смазок и др.
* * *
КРЕМНИЙОРГАНИЧЕСКИЕ ЖИДКОСТИ — КРЕМНИЙОРГАНИ́ЧЕСКИЕ ЖИ́ДКОСТИ (силиконовые масла), один из видов кремнийорганических полимеров. Применяются в качестве гидравлических жидкостей, гидрофобизаторов, смазок и др.
БОЛЬШОЙ ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
КРЕМНИЙОРГАНИЧЕСКИЕ ЖИДКОСТИ (силиконовые масла) — один из видов кремнийорганических полимеров. Применяются в качестве гидравлических жидкостей, гидрофобизаторов, смазок и др.
ПОЛЕЗНЫЕ СЕРВИСЫ
кремнийорганические каучуки
ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
Кремнийоргани́ческие каучу́ки́ (силиконовые каучуки), один из видов кремнийорганических полимеров невысокой молекулярной массы. Применяются в производстве оболочек проводов и кабелей, трубок для переливания крови, протезов (например, искусственных клапанов сердца) и др. Жидкие кремнийорганические каучуки — герметики.
* * *
КРЕМНИЙОРГАНИЧЕСКИЕ КАУЧУКИ — КРЕМНИЙОРГАНИ́ЧЕСКИЕ КАУЧУ́КИ (силиконовые каучуки), один из видов кремнийорганических полимеров невысокой молекулярной массы. Применяются в производстве оболочек проводов и кабелей, трубок для переливания крови, протезов (напр., искусственных клапанов сердца) и др. Жидкие кремнийорганические каучуки — герметики.
БОЛЬШОЙ ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
КРЕМНИЙОРГАНИЧЕСКИЕ КАУЧУКИ (силиконовые каучуки) — один из видов кремнийорганических полимеров невысокой молекулярной массы. Применяются в производстве оболочек проводов и кабелей, трубок для переливания крови, протезов (напр., искусственных клапанов сердца) и др. Жидкие кремнийорганические каучуки — герметики.
ПОЛЕЗНЫЕ СЕРВИСЫ
кремнийорганические пластики
ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
Кремнийоргани́ческие пла́стики — один из видов кремнийорганических полимеров. Применяются в производстве электроизоляционных лаков, компаундов, клеёв, стеклопластиков и др.
* * *
КРЕМНИЙОРГАНИЧЕСКИЕ ПЛАСТИКИ — КРЕМНИЙОРГАНИ́ЧЕСКИЕ ПЛА́СТИКИ, один из видов кремнийорганических полимеров. Применяются в производстве электроизоляционных лаков, компаундов, клеев, стеклопластиков и др.
БОЛЬШОЙ ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
КРЕМНИЙОРГАНИЧЕСКИЕ ПЛАСТИКИ — один из видов кремнийорганических полимеров. Применяются в производстве электроизоляционных лаков, компаундов, клеев, стеклопластиков и др.
ПОЛЕЗНЫЕ СЕРВИСЫ
кремнийорганические полимеры
ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
Кремнийоргани́ческие полиме́ры — синтетические полимеры, в молекулах которых содержатся атомы кремния и углерода. Наибольшее значение в промышленности имеют полиорганосилоксаны (полисилоксаны, силиконы), основная молекулярная цепь которых построена из чередующихся атомов кремния и кислорода, а атомы углерода входят в состав боковых (обрамляющих) групп, связанных с атомом кремния:
HO[-Si(R, R’)-О-Si(R, R’)-О-]nH
(R, R’ — органические радикалы, например СН3-). В зависимости от молекулярной массы кремнийорганические полимеры — вязкие бесцветные жидкости (кремнийорганические жидкости), твердые эластичные вещества (кремнийорганические каучуки) или хрупкие продукты (кремнийорганические пластики). Наиболее важные свойства кремнийорганических полимеров — хорошие диэлектрические характеристики, высокая термостойкость, гидрофобность, физиологическая инертность; некоторые каучуки морозостойки.
* * *
КРЕМНИЙОРГАНИЧЕСКИЕ ПОЛИМЕРЫ — КРЕМНИЙОРГАНИ́ЧЕСКИЕ ПОЛИМЕ́РЫ (силиконы), синтетические полимеры, в молекулах которых содержатся атомы кремния и углерода. Наибольшее значение в промышленности имеют полиорганосилоксаны (полисилоксаны), основная молекулярная цепь которых построена из чередующихся атомов кремния и кислорода, а атомы углерода входят в состав боковых (обрамляющих) групп, связанных с атомом кремния:
НО[ — Si(R,R») — O — Si(R, R») — O — ]nH (R, R» — органические радикалы, напр. СН3 -). В зависимости от молекулярной массы кремнийорганические полимеры — вязкие бесцветные жидкости (кремнийорганические жидкости), твердые эластичные вещества (кремнийорганические каучуки) или хрупкие продукты (кремнийорганические пластики). Наиболее важные свойства кремнийорганических полимеров — хорошие диэлектрические характеристики, высокая термостойкость, гидрофобность, физиологическая инертность; некоторые каучуки морозостойки.
БОЛЬШОЙ ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
КРЕМНИЙОРГАНИЧЕСКИЕ полимеры (силиконы) — синтетические полимеры, в молекулах которых содержатся атомы кремния и углерода. Наибольшее значение в промышленности имеют полиорганосилоксаны (полисилоксаны), основная молекулярная цепь которых построена из чередующихся атомов кремния и кислорода, а атомы углерода входят в состав боковых (обрамляющих) групп, связанных с атомом кремния:НОКРЕМНИЙОРГАНИЧЕСКИЕ соединения — соединения, содержащие в молекуле атом кремния, связанный с атомом углерода непосредственно или через атомы других элементов (O, N, S и др.). Известны, напр., органогалогенсиланы RnSiX4-n, органосиланы R4Si, органосилоксаны R3SiOSiR3 и др., где R — органический радикал, Х — галоген. Применяются для получения кремнийорганических жидкостей, каучуков, клеев, лаков.
ЭНЦИКЛОПЕДИЯ КОЛЬЕРА
КРЕМНИЙОРГАНИЧЕСКИЕ ПОЛИМЕРЫ — силиконы, представляют собой большую группу разнообразных жидкостей, каучуков и смол. Все они содержат кремний, связанный с органическим углеродом непосредственно или через кислород (полиорганосилоксаны). Кремнийорганические полимерные жидкости не имеют запаха, сильно различаются по вязкости, температуре кипения и замерзания. Они очень термостойки и если горят, то с большим трудом, мало подвержены воздействию воды, большинства химических и физических факторов, разрушающих обычные органические материалы. В свою очередь, и они очень мало влияют или не влияют совсем на большинство таких органических материалов, как пластмассы, каучуки, краски или живые ткани и организмы. Кремнийорганические жидкости являются хорошими электроизоляционными материалами, прозрачны и обладают гидрофобными свойствами. Такое редкое сочетание физических свойств позволяет использовать их в присадках для моторных масел, для изготовления различных смазочных веществ, гидравлических и демпферных жидкостей, используемых в широком диапазоне положительных и отрицательных температур, в кулинарии в составе варенья и джемов (для предупреждения вспенивания), в косметике, лакокрасочных покрытиях, для пропитки одежды и обивочных тканей, в пленках, покрывающих стенки сосудов для хранения некоторых жидких лекарств, чувствительных к контакту со стеклянной поверхностью, в составе мебельных и автомобильных полиролей, медицинском оборудовании, производстве асфальта и т.д. Тонкие пленки, оставляемые после обработки поверхности кремнийорганическими полиролями и пропитанными ими полировальными тканями, обладают исключительными пыле- и водоотталкивающими свойствами. Поверхность после такой обработки не смачивается водой и легко очищается от грязи. Кремнийорганические полимерные жидкости используются и в чистом виде. Точность чувствительных приборов и устойчивость их к повреждениям часто повышаются, если в качестве амортизирующих жидкостей применяются кремнийорганические полимеры. Хорошо подобранная жидкость устраняет нежелательное дрожание и скачки стрелки, даже если прибор испытывает значительные вибрации. Кремнийорганические жидкости позволяют снять вибрацию маховиков в двигателях различных типов от автомобильных моторов до локомотивных дизелей. Кремнийорганические полимеры обладают хорошей сжимаемостью, что дает возможность применять их в жидкостных амортизаторах самолетных шасси. Поскольку большинство органических материалов не прилипает к кремнийорганическим полимерам, кремнийорганические жидкости часто используют в виде пленок, чтобы облегчить отделение готового изделия от формы (при формовании резин или пластмасс и при литье металлов под давлением). Термо- и водостойкость кремнийорганических жидкостей вместе с их отличными электроизоляционными свойствами и устойчивостью к пробою в электрических полях позволяет применять их в изоляции свечей авиадвигателей, в радио- и рентгеновском оборудовании, антеннах, переключателях, системах зажигания судовых двигателей, аккумуляторных батареях и электрических кабелях. Они также обеспечивают длительный срок и надежность работы конденсаторов и небольших трансформаторов, предназначенных для использования при высоких температурах. Жидкости, в молекулах которых к каждому атому кремния присоединены одна метильная группа CH3 и один атом водорода H

нашли широкое применение для обработки (аппретирования) текстиля. Ткани, обработанные ими, имеют дорогой вид и приятны на ощупь, к тому же приобретают водоотталкивающие свойства. На них не остается пятен от водосодержащих жидкостей — молока, безалкогольных напитков, кофе и даже чернил. Более того, силиконовый аппрет не удаляется ни стиркой, ни химической чисткой. Эти преимущества чрезвычайно ценны для одежных и обивочных тканей.
Смолы. Кремнийорганические смолы благодаря своим превосходным качествам находят разнообразное применение. Исключительная гидрофобность, термостойкость и другие ценные качества материалов на их основе позволили повысить надежность работы машин и оборудования, уменьшить их вес, сократить расход материалов и способствовали созданию новых более совершенных электроизоляторов, защитных покрытий и т.д. Ниже указаны основные области применения кремнийорганических смол. Смолы для покрытий используются в производстве красок, лаков и эмалей для улучшения внешнего вида и защиты объектов от коррозии и от воздействия высоких температур (например, в случае металлических дымовых труб). Связующие для слоистых материалов применяются для соединения в блоки большого числа слоев бумаги, ткани, асбеста или стеклоткани с целью получения прочных, надежных листовых материалов — слоистых диэлектриков, используемых для изготовления электрических панелей, изоляторов и прокладок в высоковольтных трансформаторах. Смолы для разобщающих покрытий используют там, где требуется «нелипучая» (антиадгезионная) поверхность. Примерами служат покрытия для противней в пекарнях и для вафельниц. Водоотталкивающие смолы применяют в составах для пропитки или обмазки каменной кладки и для получения водостойкого бетона. Формуемые смолы сходны со связующими для слоистых материалов с тем лишь различием, что в них вместо ткани или бумаги используются наполнители. Этим смолам можно придавать самую сложную форму. Из них штампуют втулки, шестерни, детали электрических переключателей, разъемов, патронов, электронного оборудования и моторов. Электроизоляционные материалы, сделанные из кремнийорганических смол, термостойки, устойчивы к озону и агрессивным средам. Переход на детали из таких смол позволяет улучшить технические характеристики и долговечность электрооборудования.
Эластомеры. Кремнийорганические полимеры с большими молекулярными массами после соответствующей термической обработки сшиваются поперечными связями, возникающими между их молекулами, с образованием силиконового каучука, при дальнейшей вулканизации которого получаются эластомеры, почти неотличимые от резин, получаемых из натурального каучука. В зависимости от степени сшивания можно изменять свойства (эластичность, прочность, твердость и т.п.) получаемых материалов. Силиконовые резины эластичны при растяжении и по отскоку. Их можно отформовать в листы, трубы или изделия сложной формы, а также превратить в массу, затвердевающую при комнатной температуре. Они сохраняют эластичность при достаточно низких температурах, когда обычная синтетическая резина становится хрупкой, и при довольно высоких температурах, когда обычная резина превращается в клейкую массу. Они также не подвержены старению, воздействию погоды, воды, электричества, большинства кислот, щелочей, солей и масел. Такие свойства полиорганосиликоновых эластомеров неоценимы для многих специальных целей. Неполный список изделий из них включает: прокладки и заглушки в домашних паровых утюгах и тостерах; изолирующие трубки для защиты свечей зажигания и электрооборудования в автомобилях, самолетах и судах; изоляционные втулки для конденсаторов и трансформаторов; изоляторы для наружной осветительной арматуры, электрических печей и нагревателей, моторов и навигационных систем; упругие уплотнители и замазки; покрытия для тканей из стеклянного и асбестового волокна и герметизирующих прокладок для самолетов, летающих на больших высотах (см. также КАУЧУК И РЕЗИНА).
Химические свойства. Силоксаны содержат два или более атомов кремния, связанных посредством одного или нескольких атомов кислорода:

Два атома кремния, связанные таким образом, образуют дисилоксан, три — трисилоксан; полисилоксан содержит в молекуле большое число атомов кремния. Замкнутое кольцо из атомов кремния и кислорода

образует циклосилоксан (в данном случае — циклотрисилоксан, поскольку это циклическая структура с тремя атомами кремния). К свободным связям кремния (показанным в этих примерах черточками) могут присоединяться другие атомы кислорода. Если все связи кремния присоединены к кислороду, образуя регулярную структуру, то мы имеем дело с диоксидом кремния (кремнеземом или кварцем) SiO2 — одним из наиболее распространенных соединений в земной коре. С кремнием могут быть связаны небольшие органические группы. С метильными группами (-CH3) образуются метилсилоксаны (или метилсиликоны) — очень ценные химические продукты. Если каждый атом кремния соединен с тремя метильными группами, образуется гексаметилдисилоксан:

Это летучая жидкость, внешне напоминающая бесцветный бензин. Две метильные группы присоединены к каждому атому кремния в самых ценных продуктах из всех типов промышленных силиконов — в циклических и линейных силоксанах, примерами которых могут служить октаметилциклотетрасилоксан (I) и полидиметилсилоксан (II):

Известны способы превращения циклосилоксанов в полидиметилсилоксаны, которые могут состоять из 15 000 и более диметилсилоксановых единиц. Можно не допустить образования молекул полидиметилсилоксанов столь большого размера, добавляя вещество, содержащее триметилсилоксановые единицы, чтобы оборвать рост полидиметилсилоксановой цепи при достижении ее желаемой длины. При этом получается одна из разновидностей кремнийорганических жидкостей со структурой

Вязкость таких соединений возрастает по мере увеличения n, чему соответствует переход от очень подвижных, похожих на бензин, жидкостей к более вязким маслам и, наконец, к смолообразным веществам. Если к кремнию присоединена только одна органическая группа, то возникает сетчатая структура, характерная для полисилоксановых смол:

Обычно в производимых промышленностью таких смолах R — это метильные или фенильные (C6H5) группы. Силоксаны могут быть получены сочетанием структурных единиц всех указанных типов, т.е. с одной, двумя, тремя органическими группами при кремнии или вообще без них. Органические группы могут быть одинаковыми или представлять собой комбинацию различных типов групп. Изменяя тип и число групп при кремнии, можно получить почти бесконечное разнообразие структур. В большинстве кремнийорганических полимеров такими группами обычно являются метил, фенил или их комбинация, подобранная для получения определенных свойств. Историческая справка. Созданию большого разнообразия кремнийорганических соединений, выпускаемых современной промышленностью, предшествовала работа многих химиков в течение более 150 лет. Начало положил Й.Берцелиус открытием кремния (1823) (см. КРЕМНИЙ). Он показал, что кремний воспламеняется и энергично сгорает в токе горячего газообразного хлора с образованием жидкого вещества с удушливым запахом. Это тетрахлорид кремния SiCl4 — очень реакционноспособное соединение. С водой тетрахлорид кремния легко образует диоксид кремния и соляную кислоту: SiCl4 + 2H2O -> SiO2 + 4HCl В 1844 французский химик Эбельман показал, что SiCl4 реагирует со спиртом, образуя приятно пахнущую жидкость — тетраэтилортосиликат (тетраэтоксисилан), применяемый в наше время в больших количествах в производстве кремнийорганических полимеров: SiCl4 + 4C2H5OH -> Si(OC2H5)4 + 4HCl В 1857 Ф. Велер нагрел кремний с хлороводородом и получил дымящую жидкость — трихлорсилан HSiCl3, еще один важный промежуточный продукт для производства кремнийорганических полимеров. Ш. Фридель, профессор Сорбонны, и Дж. Крафтс, студент из Бостона, обучавшийся в Париже, сообщили в 1863, что ими получено соединение, в котором органический радикал присоединен непосредственно к кремнию, и поэтому считается, что именно эти исследователи осуществили самый важный синтез в истории кремнийорганических соединений. Использованный ими метод в наше время сочли бы трудоемким, но он привел к успеху. Они приготовили воспламеняющееся на воздухе жидкое соединение цинка, диэтилцинк, смешали его с тетрахлоридом кремния и запаяли смесь в стеклянную трубку, которую нагревали при 160° C: 2Zn(C2H5)2 + SiCl4 -> 2ZnCl2 + Si(C2H5)4 Полученное ими новое соединение кремния — тетраэтилсилан, в противоположность любым его ранее известным жидким соединениям, оказалось очень инертно: вода, кислоты и щелочи на него не действовали. Эта работа привлекла внимание молодого немецкого химика А.Ладенбурга. Ладенбург нашел способ управления реакцией с диэтилцинком, так что стало возможным по желанию присоединять к кремнию одну, две, три или четыре этильные группы. Полученный им диэтилдиэтоксисилан (C2H5)2 Si(OC2H5)2 реагировал с водой, образуя спирт и маслянистую жидкость:

(В диэтилдиэтоксисилане этильные группы, присоединенные непосредственно к кремнию, действительно связаны очень прочно, но этоксильные группы легко удаляются водой c образованием спирта.) Полученная жидкость разлагалась только при очень высоких температурах и не затвердевала при температурах много ниже точки замерзания воды. Так в 1872 Ладенбург синтезировал предшественник современных промышленных кремнийорганических полимеров, но потребовалось много усовершенствований, прежде чем стало возможным развитие промышленности кремнийорганических полимеров. Заметный вклад в исследование кремнийорганических соединений в период 1898-1939 внес Ф. Киппинг из Ноттингемского университета в Англии. В конце 1930-х годов лишь немногие химики осознали огромную потенциальную ценность полисилоксанов. Среди них выделялись Дж. Хайд («Стекольные заводы Корнинга») и Р. Макгрегор из Института Меллона в США и К. А. Андрианов в России. В 1945 Ю. Рохов обнаружил, что пары органических хлоридов реагируют с нагретым кремнием, образуя органохлорсиланы. Процесс наиболее гладко протекает с метилхлоридом. В идеальном случае реакция описывается следующим уравнением: 2CH3Cl + Si -> (CH3)2SiCl2 Процессом можно управлять, благоприятствуя этой реакции, но во всех случаях образуются побочные продукты CH3SiCl3, (CH3)3SiCl, SiCl4, HSiCl3, CH3SiHCl2, Si2Cl6 и многие другие соединения. Почти все они могут быть использованы. Для разделения продуктов смесь перегоняют, а полученные вещества применяют для синтеза разнообразных кремнийорганических полимеров. Процесс удобен для крупномасштабного производства кремнийорганических соединений. Это открытие вызвало новый взрыв интереса к химии и технологии кремнийорганических полимеров. Вскоре был открыт другой замечательный процесс, использующий дешевые углеводороды и трихлорид бора в качестве катализатора. Это позволило снизить стоимость производства целого спектра кремнийорганических соединений и цену товарных продуктов. Два примера этого процесса приведены ниже:

При обработке водой триметилхлорсилана происходит его гидролиз и получается одна из простейших промышленных кремнийорганических жидкостей, гексаметилдисилоксан: 2(CH3)3SiCl + H2O -> (CH3)3Si-O-Si(CH3)3 + 2HCl Гидролиз смеси триметилхлорсилана и диметилдихлорсилана, ведущий к более сложному соединению, представлен уравнением

В присутствии избытка диметилдихлорсилана образуются полимеры уже упоминавшегося типа:

ЛИТЕРАТУРА
Соболевский М.В. и др. Свойства и области применения кремнийорганических продуктов. М., 1975 Хананашвили Л.М., Андрианов К.А. Технология элементоорганических мономеров и полимеров. М., 1983
ПОЛЕЗНЫЕ СЕРВИСЫ
кремнийорганические соединения
ЭНЦИКЛОПЕДИЧЕСКИЙ СЛОВАРЬ
Кремнийоргани́ческие соедине́ния — соединения, содержащие в молекуле атом кремния, связанный с атомом углерода непосредственно или через атомы других элементов (О, N, S и др.). Известны, например, органогалогенсиланы RnSiX4-n, органосиланы R4Si, органосилоксаны R3SiOSiR3 и др., где R — органический радикал, Х — галоген. Применяются для получения кремнийорганических жидкостей, каучуков, клеёв, лаков.
* * *
КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — КРЕМНИЙОРГАНИ́ЧЕСКИЕ СОЕДИНЕ́НИЯ, соединения, содержащие в молекуле атом кремния, связанный с атомом углерода непосредственно или через атомы других элементов (O, N, S и др.). Известны, напр., органогалогенсиланы RnSiX4-n, органосиланы R4Si, органосилоксаны R3SiOSiR3 и др., где R — органический радикал, Х — галоген. Применяются для получения кремнийорганических жидкостей, каучуков, клеев, лаков.
ПОЛЕЗНЫЕ СЕРВИСЫ
кремнийорганические эмали
ПРАКТИЧЕСКИЙ ТОЛКОВЫЙ СЛОВАРЬ
Кремнийорганические эмали представляют собой взвеси пигментов и наполнителей в кремнийорганическом лаке, в который добавлен растворитель. Эти эмали можно использовать для окраски практически любых поверхностей — они хорошо держатся на бетоне, штукатурке, металле, картоне, дереве, пластмассах, стекле и бумаге. Кремнийорганические эмали щелочестойкие, ими можно красить по свежей штукатурке и бетону; их можно применять и для наружных и для внутренних работ. Эмали хорошо выдерживают перепады температур, защищают металлические поверхности от коррозии (их рекомендуется использовать для окраски радиаторов отопления, труб и арматуры).
Поверхность под кремнийорганические эмали готовят так же, как под алкидные краски. В некоторых случаях можно обойтись без грунтования и наносить эмаль сразу после очистки поверхности. Кремнийорганические эмали отличаются хорошим блеском и чистотой тона.
Через некоторое время на лакокрасочном покрытии появляются сколы, вмятины, трещины, царапины, повреждается грунтовка и, что особенно опасно, обнажается металл. Всякое «ранение» металла ведет к его ржавлению, которое неизбежно будет захватывать все новые и новые участки. Подкраска требуется и тогда, когда предыдущее лакокрасочное покрытие просто состарилось, «потеряло вид». Небольшие дефекты лакокрасочного покрытия, протяженностью менее 5 мм, закрасить «незаметно» довольно просто. Это лучше всего сделать с помощью распылителя, приняв меры предосторожности от загрязнения свежего покрытия пылью, приносимой ветром, и использовать для этой цели (из тех же соображений) быстросохнущие эмали, например нитроэмали.
ПОЛЕЗНЫЕ СЕРВИСЫ
кремнийорганический
СЛИТНО. РАЗДЕЛЬНО. ЧЕРЕЗ ДЕФИС
ОРФОГРАФИЧЕСКИЙ СЛОВАРЬ
СИНОНИМЫ
прил., кол-во синонимов: 2
силиконовый, кремнеорганический
МОРФЕМНО-ОРФОГРАФИЧЕСКИЙ СЛОВАРЬ
кремн/ий/орган/и́ческ/ий.
ПОЛЕЗНЫЕ СЕРВИСЫ

Химические и физические свойства
Согласно данным британского Королевского химического общества, кремний является седьмым по распространенности элементом во вселенной и вторым на Земле после кислорода. Кремний (силициум) составляет около 25 процентов земной коры и является очень полезным элементом, жизненно важным для многих отраслей промышленности.
Химические и физические свойства:

- Атомный номер (количество протонов в ядре): 14.
- На внешнем энергетическом уровне вещества находится 4 электрона.
- Атомный символ (в периодической таблице элементов): Si.
- Плотность: 2,33296 грамма на кубический сантиметр.
- Атомный вес (молярная масса атома): 28.09.
- Состояние при комнатной температуре: твердое.
- Точка кипения: 3265 градусов Цельсия (5,909 градуса по Фаренгейту).
- Количество изотопов (атомов одного и того же элемента с разным количеством нейтронов): 24.
- Температура плавления: 1414 градусов по Цельсию (2577 градусов по Фаренгейту).
- Наиболее распространенный изотоп: Si-28 (природное содержание — 92%).
- Электронная формула кремния — 1s 22s 22p 63s 23p2.
- Параметр кристаллической решетки кремния равен 0,54307 нм.
- Является 14-м элементом таблицы Менделеева.
В природе кремний не встречается сам по себе. Обычно он связывается с двумя молекулами кислорода, в результате чего обнаруживается в виде диоксида кремния, иначе называемого кремнеземом, из которого состоит кварц.
Качества вещества

Силиций был впервые выделен в 1824 году шведским химиком Йенсом Якобом Берцелиусом, который также обнаружил церий, селен и торий, согласно Фонду химического наследия. Ученый нагревал кремнезем с калием, чтобы очистить кремний. Сегодня процесс получения выглядит иначе — происходит нагревание углерода с кремнеземом в форме песка, чтобы изолировать элемент.
Кремний не является ни металлом, ни неметаллом: это металлоид, элемент, который находится где-то посередине. Эта категория — серая область, нет четкого определения того, каким требованиям она отвечает. Но металлоиды обычно имеют свойства как металлов, так и неметаллов. Кремний является полупроводником, это означает, что он проводит электричество. Однако, в отличие от типичного металла, он лучше проводит ток при повышении температуры (металлы же ухудшают проводимость повышенных температурах).

Кремний находит применение и в производстве низкотехнологичных творений, таких как кирпичи и керамика. При присоединении водорода к оксиду силиция образуется гидроксид. Гидрозоли кремниевых кислот применяют в фототехнике, а также в качестве адсорбентов. Но высокотехнологичные изделия — сфера, где элемент действительно оставляет видимый след. В качестве полупроводника кремний используется для изготовления транзисторов, которые усиливают или переключают электрические токи и являются основой электроники от радиоприемников до смартфонов.
Кремний по-разному используется в солнечных элементах и компьютерных микросхемах, одним из примеров является полевой транзистор с оксидом металла и полупроводника, основной переключатель во многих электронных устройствах. Создавая пространства из чистого силиция, инженеры получают зазор, в котором электроны не могут течь — это как переключатель в положении «выключено».
Для включения металлическая пластина, подключенная к источнику питания, помещается рядом с кристаллом. Когда электричество течет, пластина становится положительно заряженной.
Электроны с отрицательным зарядом притягиваются к положительному заряду, что позволяет им совершать прыжок через сегмент чистого кремния. (Другие полупроводники, кроме кремния, также могут использоваться в транзисторах.)
Кремниевые формы жизни
Группа IV Периодической таблицы элементов содержит углерод C, силициум (кремний) Si и несколько тяжелых металлов. Это значит, что степень окисления кремния равна +4. Углерод, конечно, является строительным материалом жизни, какой мы ее знаем. Но теоретики альтернативной биохимии предполагают, что населенная живыми существами планета может существовать в какой-то другой солнечной системе. У такой формы жизни кремний якобы может заменять углерод.
Многие научно-фантастические произведения рассказывают о кремниевых формах жизни — чувствительные кристаллы, живые золотые песчинки и даже существа, чьими следами являются кирпичи из кремнезема. Это интересно, но неправильно. Кремний может превратиться во множество реалистичных структур, но его химические свойства делают маловероятной возможность стать основой для инопланетных форм жизни.

Действительно, углерод и силиций имеют много общих характеристик. Валентность кремния равна четырем, это означает, что отдельные атомы образуют четыре связи с другими элементами при образовании химических соединений. И силиций, и углерод связываются с кислородом. Оба образуют длинные цепи, называемые полимерами. В простейшем случае углерод дает полимер под названием полиацеталь, пластик, используемый в синтетических волокнах и оборудовании. Кремний дает полимерные силиконы, которые используются для водонепроницаемой ткани или смазки металлических и пластиковых деталей.
Но когда углерод окисляется — или соединяется с кислородом, скажем, во время горения — он становится газообразным углекислым газом; силиций же окисляется до твердого диоксида, называемого кремнеземом. Кремний окисляется до твердого вещества. Это одна из основных причин того, почему он не может поддерживать жизнь.

Кремнезем или песок — это твердое вещество, диоксид кремния образует решетку, в которой один атом силиция окружен четырьмя атомами кислорода. Силикатные соединения, содержащие SiO4−4, также существуют в ряде минералов — полевых шпатах, слюде, цеолитах, тальке. И эти твердые системы создают проблемы в том, что касается утилизации энергии для живой системы.
Жизненная форма нуждается в некотором способе сбора, хранения и использования энергии. Она должна исходить из окружающей среды. После поглощения энергия должна высвобождаться именно там, где это необходимо, и в то время, когда это нужно. В противном случае вся она может высвобождать тепло одновременно, сжигая жизненную форму.
В мире на основе углерода основным элементом накопления являются углеводы, которые окисляются до воды и углекислого газа. Форма жизни на основе углерода «сжигает» энергию контролируемыми шагами, используя регуляторы скорости, называемые ферментами.
Интересные факты о кремнии
Этот элемент чрезвычайно распространен и очень широко используется. Но есть вещи, которые про него знают далеко не все:


- Когда астронавты Аполлона-11 приземлились на Луну в 1969 году, они оставили белый мешочек с кремниевым диском. Микроскопическим шрифтом там написано 73 сообщения на разных языках. Они выражают пожелания мира.
- Кремний может быть опасным. При вдыхании в течение длительных периодов времени это может вызвать заболевание легких, известное как силикоз.
- Переливчатость опала — результат присутствия кремния. Этот драгоценный камень — форма кремнезема, связанная с молекулами воды.
- Карбид кремния (SiC) почти такой же твердый, как алмаз. Он оценивается 9−9,5 по шкале твердости по Моосу, лишь немного меньше, чем самое крепкое вещество, которое имеет 10 баллов твердости.
- Растения используют кремний для укрепления клеточных стенок. Элемент является важным питательным веществом, которое помогает придать устойчивость к болезням. Об этом говорится в статье 1994 года, опубликованной в журнале Proceedings.
- Кремниевая долина получила свое название от вещества, используемого в компьютерных чипах. Название это впервые появилось в 1971 году в газете Electronic news. Своя Кремниевая долина есть не только в США, но и во многих других странах.
- Аморфная форма вещества также используется в радиоэлектронной технике.
У некоторых минералы и полудрагоценных камней строение основывается на диоксиде кремния. Они различаются плотностью и цветом:

- Аметист.
- Морион.
- Цитрин.
- Горный хрусталь.
- Опал.
- Агат.
- Сердолик.
- Яшма.
И другие камни.
Современные исследования
Сегодняшние разработки в области кремниевых технологий выглядят чуть ли не фантастично: в 2006 году исследователи объявили, что создали компьютерный чип, в котором кремниевые компоненты смешаны с клетками мозга. Это было действительно сенсационное открытие. Электрические сигналы от клеток мозга могут передаваться на электронные кремниевые компоненты чипа и наоборот. Есть надежда, что со временем получится создать устройства для лечения неврологических расстройств.

Исследование 2018 года, опубликованное в журнале Nature, было посвящено тестированию нового типа устройства из кремния, квантовых компьютеров, которые могут когда-нибудь стать обычной вещью, превосходя при этом современные компьютерные технологии в возможностях выполнять большое количество вычислений параллельно. Создание этих технологий с использованием тех же методов для изготовления традиционных кремниевых чипов может ускорить разработку этих устройств. Это может привести к новому этапу использования квантовых устройств.
Ученые также обещают создать невероятно маленькие лазеры, называемые наноиглами, которые могут использоваться для передачи данных быстрее и эффективнее, чем традиционные оптические кабели.

Сверхпроводниковые лазеры выделяют тепло гораздо легче, чем стеклянные, кремниевые лазеры могут похвастаться большей мощностью, чем традиционные, так заявляет Джон Баддинг, ученый-химик из Университета Пенсильвании.
Специалисты также работают над созданием оптических волокон следующего поколения, которые объединяют сверхпроводники, а не просто стекловолокно, так написано в журнале Live Science. В радиоэлектронной технике распространено применение силана, который получают при разложении силицида магния при помощи кислоты. Традиционные кремниевые чипы изготавливаются путем нанесения слоев элемента на плоскую поверхность, обычно начиная с газа-прекурсора силан (SiH4), поэтому новые разработки оптических волокон не потребуют нового дорогостоящего оборудования.
Кре́мний (лат. Silicium), Si, химический элемент IV группы короткой формы (14-й группы длинной формы) периодической системы; атомный номер 14, атомная масса 28,0855. Природный кремний состоит из трёх стабильных изотопов: 28Si (92,2297 %), 29Si (4,6832 %), 30Si (3,0872 %). Искусственно получены радиоизотопы с массовыми числами 22–42.
Историческая справка
Широко распространённые на земле соединения кремния использовались человеком с каменного века; например, с глубокой древности до железного века кремень применяли для выделки каменных орудий труда. Переработка соединений кремния – изготовление стекла – началась в 4-м тыс. до н. э. в Древнем Египте. Элементарный кремний получен в 1824–1825 гг. Й. Берцелиусом при восстановлении фторида SiF4 металлическим калием. Новому элементу было дано названий «силиций» (от лат. silex – кремень; русское название «кремний», введённое в 1834 г. Г. И. Гессом, также происходит от слова «кремень»).
Распространённость в природе
По распространённости в земной коре кремний – второй химический элемент (после кислорода): содержание кремния в литосфере составляет 29,5 % по массе. В свободном состоянии в природе не встречается. Важнейшие минералы, содержащие кремний, – алюмосиликаты и силикаты природные (амфиболы природные, полевые шпаты, слюды и др.), а также кремнезёма минералы (кварц и другие полиморфные модификации кремния диоксида).
Свойства
Конфигурация внешней электронной оболочки атома кремния 3s23p2. В соединениях проявляет степень окисления +4, редко +1, +2, +3, –4; электроотрицательность по Полингу 1,90, потенциалы ионизации Si0→Si+→Si2+→Si3+→Si4+ соответственно равны 8,15; 16,34; 33,46 и 45,13 эВ; атомный радиус 110 пм, радиус иона Si4+ 40 пм (координационное число 4), 54 пм (координационное число 6).
Кремний – тёмно-серое твёрдое хрупкое кристаллическое вещество с металлическим блеском. Кристаллическая решётка кубическая гранецентрированная; tпл 1414 °C, tкип 2900 °C, плотность 2330 кг/м3 (при 25 °C). Теплоёмкость 20,1 Дж/(моль·К), теплопроводность 95,5 Вт/(м·К), диэлектрическая проницаемость 12; твёрдость по Моосу 7. При обычных условиях кремний – хрупкий материал; заметная пластическая деформация наблюдается при температурах выше 800 °C. Кремний прозрачен для ИК-излучения с длиной волны больше 1 мкм (коэффициент преломления 3,45 при длине волны 2–10 мкм). Диамагнитен (магнитная восприимчивость –3,9·10–6). Кремний – полупроводник, ширина запрещённой зоны 1,21 эВ (0 К); удельное электрическое сопротивление 2,3·103 Ом·м (при 25 °C), подвижность электронов 0,135–0,145, дырок – 0,048–0,050 м2/(В·с). Электрические свойства кремния очень сильно зависят от наличия примесей. Для получения монокристаллов кремния с проводимостью р-типа используют легирующие добавки B, Al, Ga, In (акцепторные примеси), с проводимостью n-типа – P, As, Sb, Bi (донорные примеси).
 Образец чистого кремния.Кремний на воздухе покрывается оксидной плёнкой, поэтому при низких температурах химически инертен; при нагревании выше 400 °C взаимодействует с кислородом (образуются оксид SiO и диоксид SiO2), галогенами (кремния галогениды), азотом (кремния нитрид Si3N4), углеродом (кремния карбид SiC) и др. Соединения кремня с водородом – силаны – получают косвенным путём. Кремний взаимодействует с металлами с образованием силицидов.
Образец чистого кремния.Кремний на воздухе покрывается оксидной плёнкой, поэтому при низких температурах химически инертен; при нагревании выше 400 °C взаимодействует с кислородом (образуются оксид SiO и диоксид SiO2), галогенами (кремния галогениды), азотом (кремния нитрид Si3N4), углеродом (кремния карбид SiC) и др. Соединения кремня с водородом – силаны – получают косвенным путём. Кремний взаимодействует с металлами с образованием силицидов.
Мелкодисперсный кремний – восстановитель: при нагревании взаимодействует с парáми воды с выделением водорода, восстанавливает оксиды металлов до свободных металлов. Кислоты-неокислители пассивируют кремний вследствие образования на его поверхности нерастворимой в кислотах оксидной плёнки. Кремний растворяется в смеси концентрированной HNO3 с HF, при этом образуется кремнефтороводородная кислота:
3Si+4HNO3+18HF=3H2[SiF6]+4NO+8H2О3Si + 4HN O_{3} + 18HF = 3H_{2}[SiF_{6}] + 4NO + 8H_{2}О.
Кремний (особенно мелкодисперсный) взаимодействует со щелочами с выделением водорода, например:
Si+2NaOH+H2O=Na2SiO3+2H2Si + 2NaOH + H_{2}O = Na_{2}SiO_{3} + 2H_{2}.
Кремний образует различные кремнийорганические соединения.
Биологическая роль
Кремний относится к микроэлементам. Суточная потребность человека в кремнии 20–50 мг (элемент необходим для правильного роста костей и соединительных тканей). В организм человека кремний попадает с пищей, а также с вдыхаемым воздухом в виде пылеобразного SiO2. При длительном вдыхании пыли, содержащей свободный SiO2, возникает силикоз.
Получение
Кремний технической чистоты (95–98 %) получают восстановлением SiO2 углеродом или металлами. Высокочистый поликристаллический кремний получают восстановлением SiCl4 или SiHCl3 водородом при температуре 1000–1100 °С, термическим разложением SiI4 или SiH4; монокристаллический кремний высокой чистоты – зонной плавкой или по методу Чохральского. Объём мирового производства кремния 2850 тыс. т/год (2020).
Применение
Кремний – основной материал микроэлектроники и полупроводниковых приборов; используется при изготовлении стёкол, прозрачных для ИК-излучения. Кремний является компонентом сплавов железа и цветных металлов (в малых концентрациях кремний повышает коррозионную стойкость и механическую прочность сплавов, улучшает их литейные свойства; в больших концентрациях может вызвать хрупкость); наибольшее значение имеют железные, медные и алюминиевые кремнийсодержащие сплавы. Кремний применяют в качестве исходного вещества для получения кремнийорганических соединений и силицидов.
Дата публикации: 19 мая 2022 г. в 17:37 (GMT+3)