ПЦР расшифровывается как «полимеразная цепная реакция». Это метод лабораторной диагностики, цель которого заключается в выявлении возбудителя инфекционного заболевания. Он является золотым стандартом в диагностике возбудителей, который был многократно проверен. ПЦР дает возможность определить патогенный микроорганизм даже в том случае, если в исследуемом материале присутствует всего несколько его молекул ДНК.

- Как работает ПЦР-анализ?
- Преимущества ПЦР-анализа
- Показания к проведению ПЦР-теста
- Типы ПЦР
- Подготовка к анализу
- Материалы для ПЦР-анализа
- Расшифровка результатов ПЦР-тестирования
Как работает ПЦР-анализ?
Практически все живые существа на планете, исключая некоторые вирусы с РНК-геномами, имеют генетический материал, представленный длинной двухцепочной молекулой ДНК. Цепочка ДНК включает нуклеотиды — небольшие молекулы с универсальным строением. Они могут идти в цепочке ДНК в разном порядке. В зависимости от этого «собираются» разные геномы.
У каждого болезнетворного микроорганизма есть свой, уникальный порядок построения цепочки ДНК, поэтому его невозможно спутать с ДНК человека.
При проведении теста ПЦР используют фрагмент чужеродного генетического материала, добытый из образца, и размножают много раз с помощью белка-фермента. За несколько часов количество копий увеличивается в несколько миллиардов раз. Это позволяет получить такое количество ДНК, которого достаточно для успешного определения его природы.
В лабораториях используют определенный набор реактивов для каждого конкретного микроорганизма, поэтому пациентам назначают не общий ПЦР-анализ, а тест на конкретный микроорганизм (например, на вирус гепатита С).
Полимеразная цепная реакция проводится качественно и количественно. Качественный тест позволяет определить, есть ли в образце генетический материал микроорганизма (вируса или бактерии), который ищет врач. Количественный тест дает информацию о том, в каком количестве содержится материал.

Преимущества ПЦР-анализа
К достоинствам этого диагностического метода относят:
- Универсальность. ПЦР-тест обнаруживает любые ДНК и РНК, даже в тех случаях, если другие диагностические методы не дали результата.
- Эффективность. Полимеразная цепная реакция позволяет диагностировать некультивируемые, трудно культивируемые и латентно существующие формы патогенных микроорганизмов.
- Высокую чувствительность. Даже если в исследуемом материале присутствует всего один фрагмент генетического материала возбудителя, тест все равно показывает его.
- Высокую специфичность метода, достигающую 100%. В ходе диагностики выявляют уникальную последовательность нуклеотидов в цепочке ДНК, характерную только для конкретного патогенного микроорганизма, провоцирующего развитие заболевания.
- Высокую скорость выполнения. Для установки реакции достаточно несколько часов. Пациент получает результаты анализа на следующий день после сдачи.
ПЦР-тест направлен на определение возбудителя заболевания, а не на реакцию на его проникновение со стороны организма.

Показания к проведению ПЦР-теста
Анализ проводят при инфекционных заболеваниях для определения природы их возбудителя. Также его назначают при:
- необходимости точно установить конкретный болезнетворный микроорганизм;
- диагностике лейкоза и других злокачественных новообразований;
- диагностике некоторых генетически обусловленных заболеваний;
- исследовании образцов биопсии, которые невозможно изучить с помощью других методов.
С помощью ПЦР-анализа выявляют различные заболевания:
- инфекционные (ВИЧ, гепатит, ТОРЧ-инфекции и т.д.);
- передающиеся половым путем (хламидии, микоплазмы, кандиды);
- внутриутробные инфекции (вирусы краснухи, герпеса, токсоплазмы);
- респираторные (в настоящее время ПЦР-тест является золотым стандартом в диагностике вируса Ковид-19);
- генетические.
С помощью ПЦР можно провести тест на определение отцовства.

Типы ПЦР
Диагностика на основе полимеразной цепной реакции имеет несколько разновидностей. Она может быть:
- инвертированной;
- с обратной транскрипцией;
- количественной;
- вложенной;
- ступенчатой.
Наиболее надежный метод — ПЦР в режиме реального времени. Это количественный или полуколичественный способ диагностики, который предусматривает постоянное наблюдение образца генетического материала датчиком. Последний регистрирует сигналы на каждом цикле реакции.
Подготовка к анализу
При подготовке к ПЦР-анализу нужно соблюдать несколько правил:
- За 10-14 дней до назначенного теста прекратить прием любых препаратов и отказаться от проведения лечебных процедур, включая физиотерапевтические. Если прием лекарственных средств жизненно необходим, об этом нужно предупредить врача заранее.
- В случае забора мазка на выявление заболеваний, передающихся половым путем, нужно в течение суток перед исследованием отказаться от спринцеваний, использования тампонов и лечебных суппозиториев, применения антисептических средств. Также в этот период необходимо воздерживаться от половых актов. За 2 часа до забора мазка нельзя мочиться.
- Если выполняется забор мазка из зева, за 3 часа до анализа нельзя чистить зубы, пить, полоскать рот (даже простой водой).
- За трое суток до сдачи материала на ПЦР-исследование нужно полностью отказаться от употребления алкоголя.
- За двое суток до проведения теста нужно исключить повышенные физические нагрузки.
Материалы для ПЦР-анализа
В зависимости от направленности теста ПЦР, в качестве материала для исследования используют:
- кровь (сыворотка или плазма);
- соскобы эпителиальных клеток (с кожных покровов, слизистой оболочки уретры, цервикального канала);
- различные биологические жидкости (околоплодная, суставная, спинномозговая жидкости, секрет предстательной железы);
- мазок из зева;
- различные биологические выделения (слюна, моча, мокрота);
- биоптат (материал, полученный путем проведения биопсии).

Расшифровка результатов ПЦР-тестирования
Результат ПЦР-анализа либо положительный, либо отрицательный. Положительный указывает на то, что у человека присутствуют фрагменты ДНК возбудителя заболевания, то есть, что человек заражен и ему необходимо лечение. Отрицательный результат ПЦР-анализа свидетельствует об отсутствии следов ДНК патогена. В таком случае человек здоров.
В некоторых случаях ПЦР-тест дает положительный результат, хотя человек не ощущает недомогания и специфических симптомов заражения. Это значит, что заболевание выявлено на ранней стадии развития. Для уточнения диагноза назначают дополнительные исследования, а затем, с учетом их результата, начинают курс лечения.
Полимеразная цепная реакция (ПЦР) – это современный инструмент диагностики разнообразных инфекций, отличающийся высокой точностью. Анализ ПЦР позволяет определять возбудителей инфекционных заболеваний, опираясь на их генетический материал (РНК или ДНК). Биологическим материалом для исследования может служить кровь, мазок из половых органов, слюна и прочее.
КОНСУЛЬТАЦИЯ ПО РЕЗУЛЬТАТАМ АНАЛИЗОВ или УЗИ — 500 руб.
Метод ПЦР был открыт в 1983. Его создатель Кэри Мюллис (США), за это открытие получил Нобелевскую премию в области химии. Уже за первые десять лет методика стала обязательным лабораторным исследованием во всем мире, получив признание ученых и врачей.

Медицинский центр Диана предлагает провести исследование по методике полимеразной цепной реакции в кратчайшие сроки и с высочайшим уровнем точности. Наша диагностическая база соответствует всем международным стандартам и требованиям.
Какие инфекции можно диагностировать методом ПЦР
На диагностику отправляют гинеколог, уролог или дерматолог. Анализ ПЦР дает возможность диагностировать большое число разнообразных инфекций. В эту группу попадают и скрытые, бессимптомные заболевания, находящиеся в инкубационном периоде:
С помощью метода ПЦР можно обнаружить:
- гепатит C, B;
- уреаплазмоз половых органов;
- ИППП – уреаплазмоз, трихомониаз, гарднереллез,;
- хламидиоз половых органов и дыхательных путей;
- микоплазмоз половых органов и дыхательных путей;
- бактериальный вагиноз;
- кандидоз половых органов;
- инфекционный мононуклеоз;
- туберкулез;
- сальмонеллез;
- туберкулез (легочные и внелегочные формы);
- листериоз, клещевые энцефалиты, болезнь Лайма;
- цитомегаловирусную, папилломавирусную, герпесную инфекцию;
- детские заразные болезни, выявляемые при беременности и до зачатия — краснуху, паротит, дифтерию, корь;
- ВИЧ.
При каждом из вышеперечисленных заболеваний, обнаруженным другим методом, желательно делать и ПЦР — исследование. Возбудители многих инфекций имеют несколько типов (штаммов), а более точная диагностика делает лечение эффективнее. Поскольку разные типы возбудителей обладают своими способами передачи инфекции, точное определение возбудителя помогает уберечь от заражения окружающих. Например, гепатитом А можно заразиться через общие предметы и посуду (болезнь грязных рук), а гепатитом С — только через кровь, медицинские инструменты и половые контакты. Это важно для тех, кто находится рядом с больным.
Суть метода полимеразной цепной реакции
Метод основан на выявлении возбудителя по участкам ДНК или РНК. ПЦР напоминает криминалистику, когда преступника находят по частичкам кожи или волосам, оставленным на месте преступления. Поскольку каждый живой организм имеет уникальную структуру ДНК или РНК. Метод позволяет безошибочно идентифицировать микроорганизм, даже если он присутствует в исследуемом материале в минимальных количествах.
ПРЦ подходит для выявления скрытого носительства, нетипичных, стертых и прочих форм заразных заболеваний. Диагностика отличается высокой точностью, поэтому практически не дает ошибок и ложных результатов. Анализ часто назначается, как дополнительное обследование, чтобы выявить конкретный тип вируса или микроба.
Достоинства и недостатки метода ПЦР
Анализ ПЦР – результативный диагностический инструмент, позволяющий врачу не только правильно определить тип инфекционного возбудителя в организме больного, но также и количество микробов. Эта особенность позволяет методу ПЦР эффективно обнаруживать и хронические инфекции, к примеру, вирусный гепатит.
Преимущества методики заключаются в следующих особенностях:
- Универсальность. Метод дает возможность определить все известные на сегодняшний день микроорганизмы, вне зависимости от вида исследуемого материала.
- Специфичность. ПЦР позволяет определить ДНК вируса или бактерии со 100% точностью, что не дает ни один другой способ диагностики.
- Чувствительность – даже один мельчайший «обломок» ДНК или РНК может быть обнаружен и идентифицирован, благодаря быстрой реакции образования копий.
- Оперативность – поставить диагноз на основании полимеразной цепной реакции можно уже через несколько часов после забора биологического материала, что дает возможность своевременно приступить к лечению заболевания.
- Возможность проведения количественного анализа. Эта особенность важна при выявлении болезней, вызванных условно-патогенной флорой (например, молочницы). Повышенное содержание грибков кандида в организме вызывает заболевание, а нормальное количество до 10 3 — 104 КОЕ/тамп. – нет.
Недостатки ПЦР-диагностики – это необходимость в высокотехнологичном оборудовании и высококвалифицированных специалистах. Для анализа нужен специальный бокс-ламинар, где поддерживается необходимая температура и обеспечивается чистота эксперимента.
Как сдать анализ ПЦР правильно: подготовка
Чтобы понять, как именно следует подготовиться к предстоящему анализу ПЦР, пациент должен обязательно уточнить у своего врача, какой именно биологический материал планируется взять. От этого непосредственно зависит подготовка.
- Выделения из половых органов, мазок из шейки матки или мочеиспускательного канала, моча – посредством исследования данных вариантов материала методом ПЦР эффективно диагностируются инфекции половых органов.
- Вирусный гепатит C, ВИЧ инфекция требуют взятие крови на анализ ПЦР.
- С помощью мазка из зева можно подтвердить или опровергнуть факт наличия инфекционного мононуклеоза.
Мазок у женщин
Для диагностики половых инфекций методом ПЦР у женщин, как правило, берут мазок из влагалища. При беременности действуют стандартные правила подготовки к анализу ПЦР.
- За пару дней до проведения ПЦР нужно отказаться от спринцеваний, средств интимной гигиены.
- За неделю до анализа на инфекции стоит перестать применять любые виды лекарственных препаратов, разумеется, если они не входят в программу подготовки к анализу ПЦР, прописанную врачом.
- За пару дней до обследования нужно начать воздерживаться от полового акта.
- Подмывание в день анализа ПЦР не нужно, гигиена половых органов проводится накануне вечером, используется только теплая вода.
- За пару часов до сдачи анализа мочиться нельзя.
Мазок у мужчин
Подготовиться к забору мазка из мочеиспускательного канала для проведения ПЦР несложно. Достаточно соблюдать изложенные ниже правила.
- За 2 суток до ПЦР обследования нужно начать воздерживаться от сексуальных отношений.
- Прием лекарств прекращается приблизительно за 7 дней до проведения анализа, если, конечно же, они не были специально назначены врачом.
- Гигиена половых органов должна проводиться накануне вечером, в день анализа ПЦР это делать не стоит.
- Рекомендуется не мочиться примерно 2-3 часа перед сдачей анализа.
Анализ крови
Если в процессе ПЦР анализа объектом изучения станет кровь пациента, правила подготовки к нему следующие.
- Оптимальное время для сдачи крови – утро, анализ нужно обязательно сдавать натощак (минимум 8 часов отсутствия пищи). Воду можно пить свободно.
- Отказ от алкоголя за сутки до ПЦР анализа, отказ от курения за час до него.
- Если здоровье допускает такой вариант, от приема лекарственных средств рекомендуется на время отказаться.
- Перед сдачей проб крови в обязательном порядке следует отдохнуть около 20 минут.
- Накануне ПЦР исследования совершенно исключены эмоциональные и физические перегрузки, они могут сказаться на результатах анализа.
Как проводится тест ПЦР?
Для исследования берется любая среда, в которой может находиться возбудитель, но чаще всего используются:
- Кровь сыворотка или плазма – для выявления множества возбудителей – вирусов гепатита B, C, D, G, герпеса, ВИЧ, цитомегаловируса;
- Моча и сок простаты – при диагностике ЗППП;
- Мокрота, плевральная жидкость, бронхоальвеолярный лаваж – для диагностики легочных и внелёгочных форм туберкулеза;
- Спинномозговая жидкость – для диагностики инфекций нервной системы;
- Околоплодная жидкость – для определения внутриутробных заражений;
- Мазок из зева – для выявления возбудителей инфекционного мононуклеоза и дифтерии;
соскобы и мазки со слизистых для диагностики ЗППП;
клетки слизистой желудка и желудочный сок — для выявления бактерии хеликобактер, вызывающей гастрит и язву.
При одном взятии материала можно обнаружить сразу нескольких возбудителей, попавших в организм. Такая универсальность избавляет пациента от дополнительных обследований и лишних трат.
Обнаружить и идентифицировать участки генетической информации удается при помощи специальных эталонных маркеров ДНК (праймеров) созданных для каждого известного возбудителя. Эталон позволяет найти «свой» фрагмент среди миллионов других. Если это происходит, запускается реакция полимеразной цепи.

ПЦР—реакция делает огромное число копий выявленного участка генной информации (репликацию). Важно, что реплицируются только участки ДНК и РНК, необходимые для проведения анализа. Поэтому так важны чистота эксперимента и умение персонала обращаться с аппаратурой и образцами.
Цепная реакция протекает очень быстро, уже через два часа участок ДНК увеличивается в миллионы раз, что позволяет обнаружить и точно идентифицировать инфекцию. Для работы с эталонами пробирки помещают в специальный прибор. При помощи программы задается алгоритм, несколько раз меняющий температуру среды и влияющий на протекание реакции. Результат ПЦР виден сразу после окончания исследования.
Как расшифровать ПЦР анализ
Проведение ПЦР исследования позволяет не просто установить тип инфекции, присутствующий в организме больного, но и оценить его с количественной точки зрения (количество микробов). Расшифровка результатов количественного анализа ПЦР играет важную роль в обнаружении и проверке эффективности лечения большого числа хронических заболеваний, скажем гепатита C.
Результатом анализа ПЦР могут стать положительный и отрицательный ответы.
- Отрицательный результат. Следы инфекции не были выявлены в биологическом материале, который являлся объектом исследования в процессе анализа ПЦР. Как правило, на основании отрицательного результата можно полагать, что в организме действительно отсутствует инфекция.
- Положительный результат. Такой результат анализа ПЦР свидетельствует о том, что следы инфекционного заболевания присутствуют в биологическом материале пациента. Положительный результат анализа ПЦР характеризуется большой степенью точности.
Иногда инфицирование выявляется на фоне полного здоровья и отсутствия признаков заболевания. Некоторые больные считают, что анализ сделан неправильно, но реальная ситуация другая. Если ПЦР показала, что возбудитель в организме есть, значит, он там действительно находится. Просто инфекционные болезни никогда не начинаются сразу после заражения, имея инкубационный период различной продолжительности.
Скрытый период может продолжаться очень долго, например, при СПИДе – несколько лет, пока какие-то механизмы не подтолкнут вирусы к размножению. Возбудитель определяется в организме носителей и тех, кто не долечился. Очень часто переходят в скрытую (латентную) форму различные ЗППП. В этом случае может понадобиться дополнительная лабораторная диагностика.
Точность расшифровки результатов ПЦР анализа
Метод ПЦР, позволяющий выявлять достаточно большое число инфекций, характеризуется повышенной точностью, чувствительностью и специфичностью. Эти характеристики говорят о том, что с помощью исследования ПЦР можно:
- Максимально точно установить отсутствие или присутствие инфекционного возбудителя.
- Максимально точно определить конкретную разновидность инфекционного возбудителя (специфичность).
- Выявить инфекцию даже в том случае, когда она имеет скрытый характер. Речь идет о низком уровне присутствия в подвергаемом исследованию биологическом материале пациента ДНК микробов (чувствительность).
Преимущества метода ПЦР над иммуноферментным анализом и прочими иммунологическими инструментами выявления инфекции заключается в том, что он реже дает ошибочные результаты. То есть расшифровка анализа ПЦР значительно реже показывает отсутствие инфекционных микробов, если они есть в организме больного.
Кроме того, почти не бывает, чтобы расшифровка анализа выявляла наличие в организме несуществующей инфекции, то есть давала ложноположительные результаты.Разумеется, стопроцентно правильными результаты ПЦР назвать все же нельзя, поэтому необходимо обязательно дополнять это исследование другими методиками.
В конце статьи см. словарь
Полимеразную цепную реакцию (ПЦР, PCR) изобрёл в 1983 году Кэри Мюллис (американский учёный). Впоследствии он получил за это изобретение Нобелевскую премию. В настоящее время ПЦР-диагностика является, одним из самых точных и чувствительных методов диагностики инфекционных заболеваний.
Полимеразная цепная реакция (ПЦР) — экспериментальный метод молекулярной биологии, способ значительного увеличения малых концентраций определённых фрагментов нуклеиновой кислоты (ДНК) в биологическом материале (пробе).
В основе метода ПЦР лежит многократное удвоение определённого участка ДНК при помощи ферментов в искусственных условиях (in vitro). В результате нарабатываются количества ДНК, достаточные для визуальной детекции. При этом происходит копирование только того участка, который удовлетворяет заданным условиям, и только в том случае, если он присутствует в исследуемом образце.
Кроме простого увеличения числа копий ДНК (этот процесс называется амплификацией), ПЦР позволяет производить множество других манипуляций с генетическим материалом (введение мутаций, сращивание фрагментов ДНК), и широко используется в биологической и медицинской практике, например, для диагностики заболеваний (наследственных, инфекционных), для установления отцовства, для клонирования генов, введения мутаций, выделения новых генов.
Специфичность и применение
ПЦР — метод молекулярной диагностики, ставший для ряда инфекций «золотым стандартом», проверен временем и тщательно апробирован клинически. Метод ПЦР позволяет определить наличие возбудителя заболевания, даже если в пробе присутствует всего несколько молекул ДНК возбудителя.
ПЦР позволяет диагностировать наличие долго растущих возбудителей, не прибегая к трудоёмким микробиологическим методам, что особенно актуально в гинекологии и урологии при диагностике урогенитальных инфекций, передающихся половым путем (ИППП).
- Исследование урогенитального тракта методом ПЦР на ИППП ;
Также, этим методом проводят диагностику вирусных инфекций, таких как гепатиты, ВИЧ, коронавирус COVID-19 и др. Чувствительность метода значительно превосходит таковую у иммунохомических и микробиологических методов, а принцип метода позволяет диагностировать наличие инфекций со значительной антигенной изменчивостью.
- тест методом ПЦР на коронавирус Covid-19, мазок из носа и зева на определение РНК вируса SARS-CoV-2;
Специфичность ПЦР при использовании технологии PCR даже для всех вирусных, хламидийных, микоплазменных, уреаплазменных и большинства других бактериальных инфекций достигает 100%. Метод ПЦР позволяет выявлять даже единичные клетки бактерий или вирусов. ПЦР-диагностика обнаруживает наличие возбудителей инфекционных заболеваний в тех случаях, когда другими методами (иммунологическими, бактериологическими, микроскопическими) это сделать невозможно.
Особенно эффективен метод ПЦР для диагностики трудно культивируемых, некультивируемых и скрыто существующих форм микроорганизмов, с которыми часто приходится сталкиваться при латентных и хронических инфекциях, поскольку этот метод позволяет избежать сложностей, связанных с выращиванием таких микроорганизмов в лабораторных условиях.
Применение ПЦР-диагностики также очень эффективно в отношении возбудителей с высокой антигенной изменчивостью и внутриклеточных паразитов. Методом ПЦР возможно выявление возбудителей не только в клиническом материале, полученном от больного, но и в материале, получаемом из объектов внешней среды (вода, почва и т. д.). В урологической и гинекологической практике — для выявления хламидиоза, уреаплазмоза, гонореи, герпеса, гарднереллёза, микоплазменной инфекции, ВПЧ — вирусов папилломы человека; в пульмонологии — для дифференциальной диагностики вирусных и бактериальных пневмоний, туберкулёза; в гастроэнтерологии — для выявления хеликобактериоза; в клинике инфекционных заболеваний — в качестве экспресс-метода диагностики сальмонеллёза, дифтерии, вирусных гепатитов В, С и G; в гематологии — для выявления цитомегаловирусной инфекции, онковирусов.
Специфичность ПЦР основана на образовании комплементарных комплексов между матрицей и праймерами — короткими синтетическими олигонуклеотидами длиной 18 — 30 букв. Каждый из праймеров сопоставим (комплементарен) с одной из цепей двуцепочечной матрицы, обрамляя начало и конец амплифицируемого участка.
После соединения (гибридизации) матрицы с праймером (отжиг), последний служит затравкой для ДНК-полимеразы при синтезе комплементарной цепи матрицы.
Проведение ПЦР
Для проведения ПЦР в простейшем случае требуются следующие компоненты:
- ДНК-матрица, содержащая тот участок ДНК, который требуется амплифицировать;
- два праймера, комплементарные концам требуемого фрагмента;
- термостабильная ДНК-полимераза;
- дезоксинуклеотидтрифосфаты (A, G, C, T);
- ионы Mg2+, необходимые для работы полимеразы;
- буферный раствор.
ПЦР проводят в амплификаторе — приборе, обеспечивающем периодическое охлаждение и нагревание пробирок, обычно с точностью не менее 0,1°C. Чтобы избежать испарения реакционной смеси, в пробирку добавляют высококипящее масло, например, вазелиновое. Добавление специфичеких ферментов может увеличить выход ПЦР-реакции.
Ход реакции
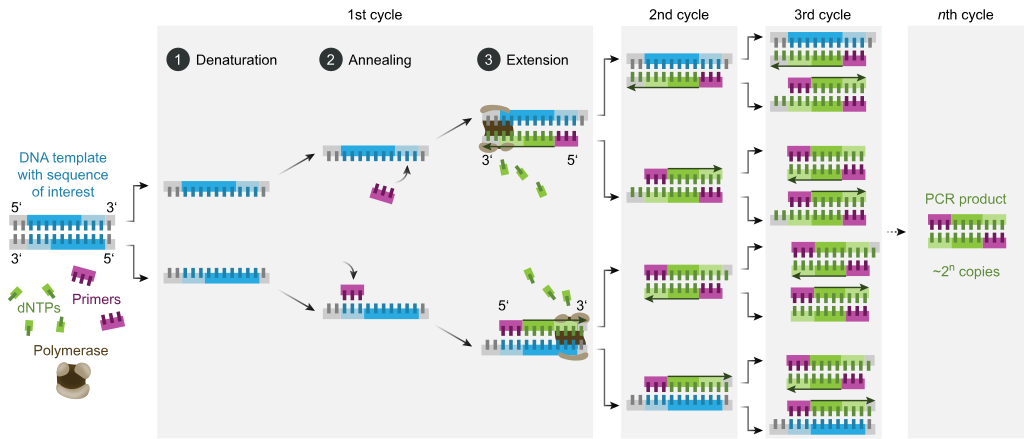
Обычно при проведении ПЦР выполняется 20 — 35 циклов, каждый из которых состоит из трех стадий. Двухцепочечную ДНК-матрицу нагревают до 94 — 96°C (или до 98°C, если используется особенно термостабильная полимераза) на 0,5 — 2 минуты, чтобы цепи ДНК разошлись. Эта стадия называется денатурацией — разрушаются водородные связи между двумя цепями. Иногда перед первым циклом проводят предварительный прогрев реакционной смеси в течение 2 — 5 минут для полной денатурации матрицы и праймеров.
Когда цепи разошлись, температуру понижают, чтобы праймеры могли связаться с одноцепочечной матрицей. Эта стадия называется отжигом. Температура отжига зависит от праймеров и обычно выбирается на 4 — 5°С ниже их температуры плавления. Время стадии — 0,5 — 2 минут.
ДНК-полимераза реплицирует матричную цепь, используя праймер в качестве затравки. Это — стадия элонгации. Температура элонгации зависит от полимеразы. Часто используемые полимеразы наиболее активны при 72°C. Время элонгации зависит как от типа ДНК-полимеразы, так и от длины амплифицируемого фрагмента. Обычно время элонгации принимают равным одной минуте на каждую тысячу пар оснований. После окончания всех циклов часто проводят дополнительную стадию финальной элонгации, чтобы достроить все одноцепочечные фрагменты. Эта стадия длится 10 — 15 мин.
Подготовка материала к исследованию и транспорт его в лабораторию
Для успешного проведения анализа важно правильно собрать материал у пациента и правильно провести его подготовку. Известно, что в лабораторной диагностике большинство ошибок (до 70%) совершается именно на этапе пробоподготовки. Для взятия крови в лаборатории ИНВИТРО в настоящее время применяются вакуумные системы, которые с одной стороны минимально травмируют пациента, а с другой — позволяют произвести взятие материала таким образом, что он не контактирует ни с персоналом, ни с окружающей средой. Это позволяет избежать контаминации (загрязнения) материала и обеспечивает объективность анализа ПЦР.
Словарь
ДНК – дезоксирибонуклеиновая кислота — биологический полимер, один из двух типов нуклеиновых кислот, обеспечивающих хранение, передачу из поколения в поколение и реализацию генетической программы развития и функционирования живых организмов. Основная роль ДНК в клетках — долговременное хранение информации о структуре РНК и белков.
РНК– рибонуклеиновая кислота — биологический полимер, близкий по своему химическому строению к ДНК. Молекула РНК построена из тех же мономерных звеньев — нуклеотидов, что и ДНК. В природе РНК, как правило, существует в виде одиночной цепочки. У некоторых вирусов РНК является носителем генетической информации. В клетке играет важную роль при передаче информации от ДНК к белку. РНК синтезируется на ДНК-матрице. Процесс этот называется транскрипцией. В ДНК имеются участки, где содержится информация, ответственная за синтез трех видов РНК, различающихся по выполняемым функциям: информационной или матричной РНК (мРНК), рибосомальной (рРНК) и транспортной (тРНК). Все три вида РНК тем или иным способом участвуют в синтезе белка. Однако информация по синтезу белка содержится только в мРНК.
Нуклеоти́ды — основная повторяющаяся единица в молекулах нуклеиновых кислот, продукт химического соединения азотистого основания, пятиуглеродного сахара (пентозы) и одной или нескольких фосфатных групп. Нуклеотиды, представленные в нуклеиновых кислотах, содержат одну фосфатную группу. Они называются по содержащемуся в них азотистому основанию — адениновый (A), содержащий аденин, гуаниновый (G) — гуанин, цитозиновый (C) — цитозин, тиминовый (Т) — тимин, урациловый (U) — урацил. В состав ДНК входят 4 типа нуклеотидов — A, T, G, C, в состав РНК также 4 типа — A, U, G, C. Сахаром в составе всех нуклеотидов ДНК является дезоксирибоза, РНК — рибоза. При образовании нуклеиновых кислот нуклеотиды, связываясь, образуют сахаро-фосфатный остов молекулы, по одну сторону которого находятся основания.
Праймер – котроткая ДНК, используемая для репликации матричной цепи. Каждый из праймеров комплементарен одной из цепей двуцепочечной матрицы, обрамляя начало и конец амплифицируемого участка.
Литература
- Глик Б., Пастернак Дж. Молекулярная биотехнология. Принципы и применение. Пер. с англ. — М.: Мир, 2002. — 589 с., илл. ISBN 5-03-003328-9
- Щелкунов С.Н. Генетическая инженерия — Новосибирск: Сиб. унив. изд-во, 2004. — 496 с.; илл. ISBN 5-94087-098-8
- Патрушев Л.И. Искусственные генетические системы — М.: Наука, 2005 — В 2 т. — ISBN 5-02-033278-X
- http://ru.wikipedia.org/wiki
ВАЖНО!
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.

A strip of eight PCR tubes, each containing a 100 μL reaction mixture

The polymerase chain reaction (PCR) is a method widely used to rapidly make millions to billions of copies (complete or partial) of a specific DNA sample, allowing scientists to take a very small sample of DNA and amplify it (or a part of it) to a large enough amount to study in detail. PCR was invented in 1983 by the American biochemist Kary Mullis at Cetus Corporation; Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA,[1] were jointly awarded the Nobel Prize in Chemistry in 1993.
PCR is fundamental to many of the procedures used in genetic testing and research, including analysis of ancient samples of DNA and identification of infectious agents. Using PCR, copies of very small amounts of DNA sequences are exponentially amplified in a series of cycles of temperature changes. PCR is now a common and often indispensable technique used in medical laboratory research for a broad variety of applications including biomedical research and criminal forensics.[2][3]
The majority of PCR methods rely on thermal cycling. Thermal cycling exposes reactants to repeated cycles of heating and cooling to permit different temperature-dependent reactions—specifically, DNA melting and enzyme-driven DNA replication. PCR employs two main reagents—primers (which are short single strand DNA fragments known as oligonucleotides that are a complementary sequence to the target DNA region) and a DNA polymerase. In the first step of PCR, the two strands of the DNA double helix are physically separated at a high temperature in a process called nucleic acid denaturation. In the second step, the temperature is lowered and the primers bind to the complementary sequences of DNA. The two DNA strands then become templates for DNA polymerase to enzymatically assemble a new DNA strand from free nucleotides, the building blocks of DNA. As PCR progresses, the DNA generated is itself used as a template for replication, setting in motion a chain reaction in which the original DNA template is exponentially amplified.
Almost all PCR applications employ a heat-stable DNA polymerase, such as Taq polymerase, an enzyme originally isolated from the thermophilic bacterium Thermus aquaticus. If the polymerase used was heat-susceptible, it would denature under the high temperatures of the denaturation step. Before the use of Taq polymerase, DNA polymerase had to be manually added every cycle, which was a tedious and costly process.[4]
Applications of the technique include DNA cloning for sequencing, gene cloning and manipulation, gene mutagenesis; construction of DNA-based phylogenies, or functional analysis of genes; diagnosis and monitoring of genetic disorders; amplification of ancient DNA;[5] analysis of genetic fingerprints for DNA profiling (for example, in forensic science and parentage testing); and detection of pathogens in nucleic acid tests for the diagnosis of infectious diseases.
Principles[edit]


PCR amplifies a specific region of a DNA strand (the DNA target). Most PCR methods amplify DNA fragments of between 0.1 and 10 kilo base pairs (kbp) in length, although some techniques allow for amplification of fragments up to 40 kbp.[6] The amount of amplified product is determined by the available substrates in the reaction, which becomes limiting as the reaction progresses.[7]
A basic PCR set-up requires several components and reagents,[8] including:
- a DNA template that contains the DNA target region to amplify
- a DNA polymerase; an enzyme that polymerizes new DNA strands; heat-resistant Taq polymerase is especially common,[9] as it is more likely to remain intact during the high-temperature DNA denaturation process
- two DNA primers that are complementary to the 3′ (three prime) ends of each of the sense and anti-sense strands of the DNA target (DNA polymerase can only bind to and elongate from a double-stranded region of DNA; without primers, there is no double-stranded initiation site at which the polymerase can bind);[10] specific primers that are complementary to the DNA target region are selected beforehand, and are often custom-made in a laboratory or purchased from commercial biochemical suppliers
- deoxynucleoside triphosphates, or dNTPs (sometimes called «deoxynucleotide triphosphates»; nucleotides containing triphosphate groups), the building blocks from which the DNA polymerase synthesizes a new DNA strand
- a buffer solution providing a suitable chemical environment for optimum activity and stability of the DNA polymerase
- bivalent cations, typically magnesium (Mg) or manganese (Mn) ions; Mg2+ is the most common, but Mn2+ can be used for PCR-mediated DNA mutagenesis, as a higher Mn2+ concentration increases the error rate during DNA synthesis;[11] and monovalent cations, typically potassium (K) ions[better source needed]
The reaction is commonly carried out in a volume of 10–200 μL in small reaction tubes (0.2–0.5 mL volumes) in a thermal cycler. The thermal cycler heats and cools the reaction tubes to achieve the temperatures required at each step of the reaction (see below). Many modern thermal cyclers make use of the Peltier effect, which permits both heating and cooling of the block holding the PCR tubes simply by reversing the electric current. Thin-walled reaction tubes permit favorable thermal conductivity to allow for rapid thermal equilibrium. Most thermal cyclers have heated lids to prevent condensation at the top of the reaction tube. Older thermal cyclers lacking a heated lid require a layer of oil on top of the reaction mixture or a ball of wax inside the tube.
Procedure[edit]
Typically, PCR consists of a series of 20–40 repeated temperature changes, called thermal cycles, with each cycle commonly consisting of two or three discrete temperature steps (see figure below). The cycling is often preceded by a single temperature step at a very high temperature (>90 °C (194 °F)), and followed by one hold at the end for final product extension or brief storage. The temperatures used and the length of time they are applied in each cycle depend on a variety of parameters, including the enzyme used for DNA synthesis, the concentration of bivalent ions and dNTPs in the reaction, and the melting temperature (Tm) of the primers.[12] The individual steps common to most PCR methods are as follows:
- Initialization: This step is only required for DNA polymerases that require heat activation by hot-start PCR.[13] It consists of heating the reaction chamber to a temperature of 94–96 °C (201–205 °F), or 98 °C (208 °F) if extremely thermostable polymerases are used, which is then held for 1–10 minutes.
- Denaturation: This step is the first regular cycling event and consists of heating the reaction chamber to 94–98 °C (201–208 °F) for 20–30 seconds. This causes DNA melting, or denaturation, of the double-stranded DNA template by breaking the hydrogen bonds between complementary bases, yielding two single-stranded DNA molecules.
- Annealing: In the next step, the reaction temperature is lowered to 50–65 °C (122–149 °F) for 20–40 seconds, allowing annealing of the primers to each of the single-stranded DNA templates. Two different primers are typically included in the reaction mixture: one for each of the two single-stranded complements containing the target region. The primers are single-stranded sequences themselves, but are much shorter than the length of the target region, complementing only very short sequences at the 3′ end of each strand.
- It is critical to determine a proper temperature for the annealing step because efficiency and specificity are strongly affected by the annealing temperature. This temperature must be low enough to allow for hybridization of the primer to the strand, but high enough for the hybridization to be specific, i.e., the primer should bind only to a perfectly complementary part of the strand, and nowhere else. If the temperature is too low, the primer may bind imperfectly. If it is too high, the primer may not bind at all. A typical annealing temperature is about 3–5 °C below the Tm of the primers used. Stable hydrogen bonds between complementary bases are formed only when the primer sequence very closely matches the template sequence. During this step, the polymerase binds to the primer-template hybrid and begins DNA formation.
- Extension/elongation: The temperature at this step depends on the DNA polymerase used; the optimum activity temperature for the thermostable DNA polymerase of Taq polymerase is approximately 75–80 °C (167–176 °F),[14][15] though a temperature of 72 °C (162 °F) is commonly used with this enzyme. In this step, the DNA polymerase synthesizes a new DNA strand complementary to the DNA template strand by adding free dNTPs from the reaction mixture that is complementary to the template in the 5′-to-3′ direction, condensing the 5′-phosphate group of the dNTPs with the 3′-hydroxy group at the end of the nascent (elongating) DNA strand. The precise time required for elongation depends both on the DNA polymerase used and on the length of the DNA target region to amplify. As a rule of thumb, at their optimal temperature, most DNA polymerases polymerize a thousand bases per minute. Under optimal conditions (i.e., if there are no limitations due to limiting substrates or reagents), at each extension/elongation step, the number of DNA target sequences is doubled. With each successive cycle, the original template strands plus all newly generated strands become template strands for the next round of elongation, leading to exponential (geometric) amplification of the specific DNA target region.
- The processes of denaturation, annealing and elongation constitute a single cycle. Multiple cycles are required to amplify the DNA target to millions of copies. The formula used to calculate the number of DNA copies formed after a given number of cycles is 2n, where n is the number of cycles. Thus, a reaction set for 30 cycles results in 230, or 1,073,741,824, copies of the original double-stranded DNA target region.
- Final elongation: This single step is optional, but is performed at a temperature of 70–74 °C (158–165 °F) (the temperature range required for optimal activity of most polymerases used in PCR) for 5–15 minutes after the last PCR cycle to ensure that any remaining single-stranded DNA is fully elongated.
- Final hold: The final step cools the reaction chamber to 4–15 °C (39–59 °F) for an indefinite time, and may be employed for short-term storage of the PCR products.

Ethidium bromide-stained PCR products after gel electrophoresis. Two sets of primers were used to amplify a target sequence from three different tissue samples. No amplification is present in sample #1; DNA bands in sample #2 and #3 indicate successful amplification of the target sequence. The gel also shows a positive control, and a DNA ladder containing DNA fragments of defined length for sizing the bands in the experimental PCRs.
To check whether the PCR successfully generated the anticipated DNA target region (also sometimes referred to as the amplimer or amplicon), agarose gel electrophoresis may be employed for size separation of the PCR products. The size of the PCR products is determined by comparison with a DNA ladder, a molecular weight marker which contains DNA fragments of known sizes, which runs on the gel alongside the PCR products.

Stages[edit]

Exponential amplification
As with other chemical reactions, the reaction rate and efficiency of PCR are affected by limiting factors. Thus, the entire PCR process can further be divided into three stages based on reaction progress:
- Exponential amplification: At every cycle, the amount of product is doubled (assuming 100% reaction efficiency). After 30 cycles, a single copy of DNA can be increased up to 1,000,000,000 (one billion) copies. In a sense, then, the replication of a discrete strand of DNA is being manipulated in a tube under controlled conditions.[16] The reaction is very sensitive: only minute quantities of DNA must be present.
- Leveling off stage: The reaction slows as the DNA polymerase loses activity and as consumption of reagents, such as dNTPs and primers, causes them to become more limited.
- Plateau: No more product accumulates due to exhaustion of reagents and enzyme.
Optimization[edit]
In practice, PCR can fail for various reasons, such as sensitivity or contamination.[17][18] Contamination with extraneous DNA can lead to spurious products and is addressed with lab protocols and procedures that separate pre-PCR mixtures from potential DNA contaminants.[8] For instance, if DNA from a crime scene is analyzed, a single DNA molecule from lab personnel could be amplified and misguide the investigation. Hence the PCR-setup areas is separated from the analysis or purification of other PCR products, disposable plasticware used, and the work surface between reaction setups needs to be thoroughly cleaned.
Specificity can be adjusted by experimental conditions so that no spurious products are generated. Primer-design techniques are important in improving PCR product yield and in avoiding the formation of unspecific products. The usage of alternate buffer components or polymerase enzymes can help with amplification of long or otherwise problematic regions of DNA. For instance, Q5 polymerase is said to be ~280 times less error-prone than Taq polymerase.[19][20] Both the running parameters (e.g. temperature and duration of cycles), or the addition of reagents, such as formamide, may increase the specificity and yield of PCR.[21] Computer simulations of theoretical PCR results (Electronic PCR) may be performed to assist in primer design.[22]
Applications[edit]
Selective DNA isolation[edit]
PCR allows isolation of DNA fragments from genomic DNA by selective amplification of a specific region of DNA. This use of PCR augments many ways, such as generating hybridization probes for Southern or northern hybridization and DNA cloning, which require larger amounts of DNA, representing a specific DNA region. PCR supplies these techniques with high amounts of pure DNA, enabling analysis of DNA samples even from very small amounts of starting material.
Other applications of PCR include DNA sequencing to determine unknown PCR-amplified sequences in which one of the amplification primers may be used in Sanger sequencing, isolation of a DNA sequence to expedite recombinant DNA technologies involving the insertion of a DNA sequence into a plasmid, phage, or cosmid (depending on size) or the genetic material of another organism. Bacterial colonies (such as E. coli) can be rapidly screened by PCR for correct DNA vector constructs.[23] PCR may also be used for genetic fingerprinting; a forensic technique used to identify a person or organism by comparing experimental DNAs through different PCR-based methods.

Electrophoresis of PCR-amplified DNA fragments:
- Father
- Child
- Mother
The child has inherited some, but not all, of the fingerprints of each of its parents, giving it a new, unique fingerprint.
Some PCR fingerprint methods have high discriminative power and can be used to identify genetic relationships between individuals, such as parent-child or between siblings, and are used in paternity testing (Fig. 4). This technique may also be used to determine evolutionary relationships among organisms when certain molecular clocks are used (i.e. the 16S rRNA and recA genes of microorganisms).[24]
Amplification and quantification of DNA[edit]
Because PCR amplifies the regions of DNA that it targets, PCR can be used to analyze extremely small amounts of sample. This is often critical for forensic analysis, when only a trace amount of DNA is available as evidence. PCR may also be used in the analysis of ancient DNA that is tens of thousands of years old. These PCR-based techniques have been successfully used on animals, such as a forty-thousand-year-old mammoth, and also on human DNA, in applications ranging from the analysis of Egyptian mummies to the identification of a Russian tsar and the body of English king Richard III.[25]
Quantitative PCR or Real Time PCR (qPCR,[26] not to be confused with RT-PCR) methods allow the estimation of the amount of a given sequence present in a sample—a technique often applied to quantitatively determine levels of gene expression. Quantitative PCR is an established tool for DNA quantification that measures the accumulation of DNA product after each round of PCR amplification.
qPCR allows the quantification and detection of a specific DNA sequence in real time since it measures concentration while the synthesis process is taking place. There are two methods for simultaneous detection and quantification. The first method consists of using fluorescent dyes that are retained nonspecifically in between the double strands. The second method involves probes that code for specific sequences and are fluorescently labeled. Detection of DNA using these methods can only be seen after the hybridization of probes with its complementary DNA (cDNA) takes place. An interesting technique combination is real-time PCR and reverse transcription. This sophisticated technique, called RT-qPCR, allows for the quantification of a small quantity of RNA. Through this combined technique, mRNA is converted to cDNA, which is further quantified using qPCR. This technique lowers the possibility of error at the end point of PCR,[27] increasing chances for detection of genes associated with genetic diseases such as cancer.[5] Laboratories use RT-qPCR for the purpose of sensitively measuring gene regulation. The mathematical foundations for the reliable quantification of the PCR[28] and RT-qPCR[29] facilitate the implementation of accurate fitting procedures of experimental data in research, medical, diagnostic and infectious disease applications.[30][31][32][33]
Medical and diagnostic applications[edit]
Prospective parents can be tested for being genetic carriers, or their children might be tested for actually being affected by a disease.[2] DNA samples for prenatal testing can be obtained by amniocentesis, chorionic villus sampling, or even by the analysis of rare fetal cells circulating in the mother’s bloodstream. PCR analysis is also essential to preimplantation genetic diagnosis, where individual cells of a developing embryo are tested for mutations.
- PCR can also be used as part of a sensitive test for tissue typing, vital to organ transplantation. As of 2008, there is even a proposal to replace the traditional antibody-based tests for blood type with PCR-based tests.[34]
- Many forms of cancer involve alterations to oncogenes. By using PCR-based tests to study these mutations, therapy regimens can sometimes be individually customized to a patient. PCR permits early diagnosis of malignant diseases such as leukemia and lymphomas, which is currently the highest-developed in cancer research and is already being used routinely. PCR assays can be performed directly on genomic DNA samples to detect translocation-specific malignant cells at a sensitivity that is at least 10,000 fold higher than that of other methods.[35] PCR is very useful in the medical field since it allows for the isolation and amplification of tumor suppressors. Quantitative PCR for example, can be used to quantify and analyze single cells, as well as recognize DNA, mRNA and protein confirmations and combinations.[27]
Infectious disease applications[edit]
PCR allows for rapid and highly specific diagnosis of infectious diseases, including those caused by bacteria or viruses.[36] PCR also permits identification of non-cultivatable or slow-growing microorganisms such as mycobacteria, anaerobic bacteria, or viruses from tissue culture assays and animal models. The basis for PCR diagnostic applications in microbiology is the detection of infectious agents and the discrimination of non-pathogenic from pathogenic strains by virtue of specific genes.[36][37]
Characterization and detection of infectious disease organisms have been revolutionized by PCR in the following ways:
- The human immunodeficiency virus (or HIV), is a difficult target to find and eradicate. The earliest tests for infection relied on the presence of antibodies to the virus circulating in the bloodstream. However, antibodies don’t appear until many weeks after infection, maternal antibodies mask the infection of a newborn, and therapeutic agents to fight the infection don’t affect the antibodies. PCR tests have been developed that can detect as little as one viral genome among the DNA of over 50,000 host cells.[38] Infections can be detected earlier, donated blood can be screened directly for the virus, newborns can be immediately tested for infection, and the effects of antiviral treatments can be quantified.
- Some disease organisms, such as that for tuberculosis, are difficult to sample from patients and slow to be grown in the laboratory. PCR-based tests have allowed detection of small numbers of disease organisms (both live or dead), in convenient samples. Detailed genetic analysis can also be used to detect antibiotic resistance, allowing immediate and effective therapy. The effects of therapy can also be immediately evaluated.
- The spread of a disease organism through populations of domestic or wild animals can be monitored by PCR testing. In many cases, the appearance of new virulent sub-types can be detected and monitored. The sub-types of an organism that were responsible for earlier epidemics can also be determined by PCR analysis.
- Viral DNA can be detected by PCR. The primers used must be specific to the targeted sequences in the DNA of a virus, and PCR can be used for diagnostic analyses or DNA sequencing of the viral genome. The high sensitivity of PCR permits virus detection soon after infection and even before the onset of disease.[36] Such early detection may give physicians a significant lead time in treatment. The amount of virus («viral load») in a patient can also be quantified by PCR-based DNA quantitation techniques (see below). A variant of PCR (RT-PCR) is used for detecting viral RNA rather than DNA: in this test the enzyme reverse transcriptase is used to generate a DNA sequence which matches the viral RNA; this DNA is then amplified as per the usual PCR method. RT-PCR is widely used to detect the SARS-CoV-2 viral genome.[39]
- Diseases such as pertussis (or whooping cough) are caused by the bacteria Bordetella pertussis. This bacteria is marked by a serious acute respiratory infection that affects various animals and humans and has led to the deaths of many young children. The pertussis toxin is a protein exotoxin that binds to cell receptors by two dimers and reacts with different cell types such as T lymphocytes which play a role in cell immunity.[40] PCR is an important testing tool that can detect sequences within the gene for the pertussis toxin. Because PCR has a high sensitivity for the toxin and a rapid turnaround time, it is very efficient for diagnosing pertussis when compared to culture.[41]
Forensic applications[edit]
The development of PCR-based genetic (or DNA) fingerprinting protocols has seen widespread application in forensics:
-

DNA samples are often taken at crime scenes and analyzed by PCR.
In its most discriminating form, genetic fingerprinting can uniquely discriminate any one person from the entire population of the world. Minute samples of DNA can be isolated from a crime scene, and compared to that from suspects, or from a DNA database of earlier evidence or convicts. Simpler versions of these tests are often used to rapidly rule out suspects during a criminal investigation. Evidence from decades-old crimes can be tested, confirming or exonerating the people originally convicted.
- Forensic DNA typing has been an effective way of identifying or exonerating criminal suspects due to analysis of evidence discovered at a crime scene. The human genome has many repetitive regions that can be found within gene sequences or in non-coding regions of the genome. Specifically, up to 40% of human DNA is repetitive.[5] There are two distinct categories for these repetitive, non-coding regions in the genome. The first category is called variable number tandem repeats (VNTR), which are 10–100 base pairs long and the second category is called short tandem repeats (STR) and these consist of repeated 2–10 base pair sections. PCR is used to amplify several well-known VNTRs and STRs using primers that flank each of the repetitive regions. The sizes of the fragments obtained from any individual for each of the STRs will indicate which alleles are present. By analyzing several STRs for an individual, a set of alleles for each person will be found that statistically is likely to be unique.[5] Researchers have identified the complete sequence of the human genome. This sequence can be easily accessed through the NCBI website and is used in many real-life applications. For example, the FBI has compiled a set of DNA marker sites used for identification, and these are called the Combined DNA Index System (CODIS) DNA database.[5] Using this database enables statistical analysis to be used to determine the probability that a DNA sample will match. PCR is a very powerful and significant analytical tool to use for forensic DNA typing because researchers only need a very small amount of the target DNA to be used for analysis. For example, a single human hair with attached hair follicle has enough DNA to conduct the analysis. Similarly, a few sperm, skin samples from under the fingernails, or a small amount of blood can provide enough DNA for conclusive analysis.[5]
- Less discriminating forms of DNA fingerprinting can help in DNA paternity testing, where an individual is matched with their close relatives. DNA from unidentified human remains can be tested, and compared with that from possible parents, siblings, or children. Similar testing can be used to confirm the biological parents of an adopted (or kidnapped) child. The actual biological father of a newborn can also be confirmed (or ruled out).
- The PCR AMGX/AMGY design has been shown to not only[clarification needed] facilitate in amplifying DNA sequences from a very minuscule amount of genome. However it can also be used for real-time sex determination from forensic bone samples. This provides a powerful and effective way to determine gender in forensic cases and ancient specimens.[42]

Research applications[edit]
PCR has been applied to many areas of research in molecular genetics:
- PCR allows rapid production of short pieces of DNA, even when not more than the sequence of the two primers is known. This ability of PCR augments many methods, such as generating hybridization probes for Southern or northern blot hybridization. PCR supplies these techniques with large amounts of pure DNA, sometimes as a single strand, enabling analysis even from very small amounts of starting material.
- The task of DNA sequencing can also be assisted by PCR. Known segments of DNA can easily be produced from a patient with a genetic disease mutation. Modifications to the amplification technique can extract segments from a completely unknown genome, or can generate just a single strand of an area of interest.
- PCR has numerous applications to the more traditional process of DNA cloning. It can extract segments for insertion into a vector from a larger genome, which may be only available in small quantities. Using a single set of ‘vector primers’, it can also analyze or extract fragments that have already been inserted into vectors. Some alterations to the PCR protocol can generate mutations (general or site-directed) of an inserted fragment.
- Sequence-tagged sites is a process where PCR is used as an indicator that a particular segment of a genome is present in a particular clone. The Human Genome Project found this application vital to mapping the cosmid clones they were sequencing, and to coordinating the results from different laboratories.
- An application of PCR is the phylogenic analysis of DNA from ancient sources, such as that found in the recovered bones of Neanderthals, from frozen tissues of mammoths, or from the brain of Egyptian mummies.[16] In some cases the highly degraded DNA from these sources might be reassembled during the early stages of amplification.
- A common application of PCR is the study of patterns of gene expression. Tissues (or even individual cells) can be analyzed at different stages to see which genes have become active, or which have been switched off. This application can also use quantitative PCR to quantitate the actual levels of expression
- The ability of PCR to simultaneously amplify several loci from individual sperm[43] has greatly enhanced the more traditional task of genetic mapping by studying chromosomal crossovers after meiosis. Rare crossover events between very close loci have been directly observed by analyzing thousands of individual sperms. Similarly, unusual deletions, insertions, translocations, or inversions can be analyzed, all without having to wait (or pay) for the long and laborious processes of fertilization, embryogenesis, etc.
- Site-directed mutagenesis: PCR can be used to create mutant genes with mutations chosen by scientists at will. These mutations can be chosen in order to understand how proteins accomplish their functions, and to change or improve protein function.
Advantages[edit]
PCR has a number of advantages. It is fairly simple to understand and to use, and produces results rapidly. The technique is highly sensitive with the potential to produce millions to billions of copies of a specific product for sequencing, cloning, and analysis. qRT-PCR shares the same advantages as the PCR, with an added advantage of quantification of the synthesized product. Therefore, it has its uses to analyze alterations of gene expression levels in tumors, microbes, or other disease states.[27]
PCR is a very powerful and practical research tool. The sequencing of unknown etiologies of many diseases are being figured out by the PCR. The technique can help identify the sequence of previously unknown viruses related to those already known and thus give us a better understanding of the disease itself. If the procedure can be further simplified and sensitive non-radiometric detection systems can be developed, the PCR will assume a prominent place in the clinical laboratory for years to come.[16]
Limitations[edit]
One major limitation of PCR is that prior information about the target sequence is necessary in order to generate the primers that will allow its selective amplification.[27] This means that, typically, PCR users must know the precise sequence(s) upstream of the target region on each of the two single-stranded templates in order to ensure that the DNA polymerase properly binds to the primer-template hybrids and subsequently generates the entire target region during DNA synthesis.
Like all enzymes, DNA polymerases are also prone to error, which in turn causes mutations in the PCR fragments that are generated.[44]
Another limitation of PCR is that even the smallest amount of contaminating DNA can be amplified, resulting in misleading or ambiguous results. To minimize the chance of contamination, investigators should reserve separate rooms for reagent preparation, the PCR, and analysis of product. Reagents should be dispensed into single-use aliquots. Pipettors with disposable plungers and extra-long pipette tips should be routinely used.[16] It is moreover recommended to ensure that the lab set-up follows a unidirectional workflow. No materials or reagents used in the PCR and analysis rooms should ever be taken into the PCR preparation room without thorough decontamination.[45]
Environmental samples that contain humic acids may inhibit PCR amplification and lead to inaccurate results.
Variations[edit]
- Allele-specific PCR or The amplification refractory mutation system (ARMS): a diagnostic or cloning technique based on single-nucleotide variations (SNVs not to be confused with SNPs) (single-base differences in a patient). Any mutation involving single base change can be detected by this system. It requires prior knowledge of a DNA sequence, including differences between alleles, and uses primers whose 3′ ends encompass the SNV (base pair buffer around SNV usually incorporated).[46] PCR amplification under stringent conditions is much less efficient in the presence of a mismatch between template and primer, so successful amplification with an SNP-specific primer signals presence of the specific SNP or small deletions in a sequence.[47] See SNP genotyping for more information.
- Assembly PCR or Polymerase Cycling Assembly (PCA): artificial synthesis of long DNA sequences by performing PCR on a pool of long oligonucleotides with short overlapping segments. The oligonucleotides alternate between sense and antisense directions, and the overlapping segments determine the order of the PCR fragments, thereby selectively producing the final long DNA product.[48]
- Asymmetric PCR: preferentially amplifies one DNA strand in a double-stranded DNA template. It is used in sequencing and hybridization probing where amplification of only one of the two complementary strands is required. PCR is carried out as usual, but with a great excess of the primer for the strand targeted for amplification. Because of the slow (arithmetic) amplification later in the reaction after the limiting primer has been used up, extra cycles of PCR are required.[49] A recent modification on this process, known as Linear-After-The-Exponential-PCR (LATE-PCR), uses a limiting primer with a higher melting temperature (Tm) than the excess primer to maintain reaction efficiency as the limiting primer concentration decreases mid-reaction.[50]
- Convective PCR: a pseudo-isothermal way of performing PCR. Instead of repeatedly heating and cooling the PCR mixture, the solution is subjected to a thermal gradient. The resulting thermal instability driven convective flow automatically shuffles the PCR reagents from the hot and cold regions repeatedly enabling PCR.[51] Parameters such as thermal boundary conditions and geometry of the PCR enclosure can be optimized to yield robust and rapid PCR by harnessing the emergence of chaotic flow fields.[52] Such convective flow PCR setup significantly reduces device power requirement and operation time.
- Dial-out PCR: a highly parallel method for retrieving accurate DNA molecules for gene synthesis. A complex library of DNA molecules is modified with unique flanking tags before massively parallel sequencing. Tag-directed primers then enable the retrieval of molecules with desired sequences by PCR.[53]
- Digital PCR (dPCR): used to measure the quantity of a target DNA sequence in a DNA sample. The DNA sample is highly diluted so that after running many PCRs in parallel, some of them do not receive a single molecule of the target DNA. The target DNA concentration is calculated using the proportion of negative outcomes. Hence the name ‘digital PCR’.
- Helicase-dependent amplification: similar to traditional PCR, but uses a constant temperature rather than cycling through denaturation and annealing/extension cycles. DNA helicase, an enzyme that unwinds DNA, is used in place of thermal denaturation.[54]
- Hot start PCR: a technique that reduces non-specific amplification during the initial set up stages of the PCR. It may be performed manually by heating the reaction components to the denaturation temperature (e.g., 95 °C) before adding the polymerase.[55] Specialized enzyme systems have been developed that inhibit the polymerase’s activity at ambient temperature, either by the binding of an antibody[13][56] or by the presence of covalently bound inhibitors that dissociate only after a high-temperature activation step. Hot-start/cold-finish PCR is achieved with new hybrid polymerases that are inactive at ambient temperature and are instantly activated at elongation temperature.
- In silico PCR (digital PCR, virtual PCR, electronic PCR, e-PCR) refers to computational tools used to calculate theoretical polymerase chain reaction results using a given set of primers (probes) to amplify DNA sequences from a sequenced genome or transcriptome. In silico PCR was proposed as an educational tool for molecular biology.[57]
- Intersequence-specific PCR (ISSR): a PCR method for DNA fingerprinting that amplifies regions between simple sequence repeats to produce a unique fingerprint of amplified fragment lengths.[58]
- Inverse PCR: is commonly used to identify the flanking sequences around genomic inserts. It involves a series of DNA digestions and self ligation, resulting in known sequences at either end of the unknown sequence.[59]
- Ligation-mediated PCR: uses small DNA linkers ligated to the DNA of interest and multiple primers annealing to the DNA linkers; it has been used for DNA sequencing, genome walking, and DNA footprinting.[60]
- Methylation-specific PCR (MSP): developed by Stephen Baylin and James G. Herman at the Johns Hopkins School of Medicine,[61] and is used to detect methylation of CpG islands in genomic DNA. DNA is first treated with sodium bisulfite, which converts unmethylated cytosine bases to uracil, which is recognized by PCR primers as thymine. Two PCRs are then carried out on the modified DNA, using primer sets identical except at any CpG islands within the primer sequences. At these points, one primer set recognizes DNA with cytosines to amplify methylated DNA, and one set recognizes DNA with uracil or thymine to amplify unmethylated DNA. MSP using qPCR can also be performed to obtain quantitative rather than qualitative information about methylation.
- Miniprimer PCR: uses a thermostable polymerase (S-Tbr) that can extend from short primers («smalligos») as short as 9 or 10 nucleotides. This method permits PCR targeting to smaller primer binding regions, and is used to amplify conserved DNA sequences, such as the 16S (or eukaryotic 18S) rRNA gene.[62]
- Multiplex ligation-dependent probe amplification (MLPA): permits amplifying multiple targets with a single primer pair, thus avoiding the resolution limitations of multiplex PCR (see below).
- Multiplex-PCR: consists of multiple primer sets within a single PCR mixture to produce amplicons of varying sizes that are specific to different DNA sequences. By targeting multiple genes at once, additional information may be gained from a single test-run that otherwise would require several times the reagents and more time to perform. Annealing temperatures for each of the primer sets must be optimized to work correctly within a single reaction, and amplicon sizes. That is, their base pair length should be different enough to form distinct bands when visualized by gel electrophoresis.
- Nanoparticle-assisted PCR (nanoPCR): some nanoparticles (NPs) can enhance the efficiency of PCR (thus being called nanoPCR), and some can even outperform the original PCR enhancers. It was reported that quantum dots (QDs) can improve PCR specificity and efficiency. Single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs) are efficient in enhancing the amplification of long PCR. Carbon nanopowder (CNP) can improve the efficiency of repeated PCR and long PCR, while zinc oxide, titanium dioxide and Ag NPs were found to increase the PCR yield. Previous data indicated that non-metallic NPs retained acceptable amplification fidelity. Given that many NPs are capable of enhancing PCR efficiency, it is clear that there is likely to be great potential for nanoPCR technology improvements and product development.[63][64]
- Nested PCR: increases the specificity of DNA amplification, by reducing background due to non-specific amplification of DNA. Two sets of primers are used in two successive PCRs. In the first reaction, one pair of primers is used to generate DNA products, which besides the intended target, may still consist of non-specifically amplified DNA fragments. The product(s) are then used in a second PCR with a set of primers whose binding sites are completely or partially different from and located 3′ of each of the primers used in the first reaction. Nested PCR is often more successful in specifically amplifying long DNA fragments than conventional PCR, but it requires more detailed knowledge of the target sequences.
- Overlap-extension PCR or Splicing by overlap extension (SOEing) : a genetic engineering technique that is used to splice together two or more DNA fragments that contain complementary sequences. It is used to join DNA pieces containing genes, regulatory sequences, or mutations; the technique enables creation of specific and long DNA constructs. It can also introduce deletions, insertions or point mutations into a DNA sequence.[65][66]
- PAN-AC: uses isothermal conditions for amplification, and may be used in living cells.[67][68]
- Quantitative PCR (qPCR): used to measure the quantity of a target sequence (commonly in real-time). It quantitatively measures starting amounts of DNA, cDNA, or RNA. Quantitative PCR is commonly used to determine whether a DNA sequence is present in a sample and the number of its copies in the sample. Quantitative PCR has a very high degree of precision. Quantitative PCR methods use fluorescent dyes, such as Sybr Green, EvaGreen or fluorophore-containing DNA probes, such as TaqMan, to measure the amount of amplified product in real time. It is also sometimes abbreviated to RT-PCR (real-time PCR) but this abbreviation should be used only for reverse transcription PCR. qPCR is the appropriate contractions for quantitative PCR (real-time PCR).
- Reverse Complement PCR (RC-PCR): Allows the addition of functional domains or sequences of choice to be appended independently to either end of the generated amplicon in a single closed tube reaction. This method generates target specific primers within the reaction by the interaction of universal primers (which contain the desired sequences or domains to be appended) and RC probes.
- Reverse Transcription PCR (RT-PCR): for amplifying DNA from RNA. Reverse transcriptase reverse transcribes RNA into cDNA, which is then amplified by PCR. RT-PCR is widely used in expression profiling, to determine the expression of a gene or to identify the sequence of an RNA transcript, including transcription start and termination sites. If the genomic DNA sequence of a gene is known, RT-PCR can be used to map the location of exons and introns in the gene. The 5′ end of a gene (corresponding to the transcription start site) is typically identified by RACE-PCR (Rapid Amplification of cDNA Ends).
- RNase H-dependent PCR (rhPCR): a modification of PCR that utilizes primers with a 3′ extension block that can be removed by a thermostable RNase HII enzyme. This system reduces primer-dimers and allows for multiplexed reactions to be performed with higher numbers of primers.[69]
- Single specific primer-PCR (SSP-PCR): allows the amplification of double-stranded DNA even when the sequence information is available at one end only. This method permits amplification of genes for which only a partial sequence information is available, and allows unidirectional genome walking from known into unknown regions of the chromosome.[70]
- Solid Phase PCR: encompasses multiple meanings, including Polony Amplification (where PCR colonies are derived in a gel matrix, for example), Bridge PCR[71] (primers are covalently linked to a solid-support surface), conventional Solid Phase PCR (where Asymmetric PCR is applied in the presence of solid support bearing primer with sequence matching one of the aqueous primers) and Enhanced Solid Phase PCR[72] (where conventional Solid Phase PCR can be improved by employing high Tm and nested solid support primer with optional application of a thermal ‘step’ to favour solid support priming).
- Suicide PCR: typically used in paleogenetics or other studies where avoiding false positives and ensuring the specificity of the amplified fragment is the highest priority. It was originally described in a study to verify the presence of the microbe Yersinia pestis in dental samples obtained from 14th Century graves of people supposedly killed by the plague during the medieval Black Death epidemic.[73] The method prescribes the use of any primer combination only once in a PCR (hence the term «suicide»), which should never have been used in any positive control PCR reaction, and the primers should always target a genomic region never amplified before in the lab using this or any other set of primers. This ensures that no contaminating DNA from previous PCR reactions is present in the lab, which could otherwise generate false positives.
- Thermal asymmetric interlaced PCR (TAIL-PCR): for isolation of an unknown sequence flanking a known sequence. Within the known sequence, TAIL-PCR uses a nested pair of primers with differing annealing temperatures; a degenerate primer is used to amplify in the other direction from the unknown sequence.[74]
- Touchdown PCR (Step-down PCR): a variant of PCR that aims to reduce nonspecific background by gradually lowering the annealing temperature as PCR cycling progresses. The annealing temperature at the initial cycles is usually a few degrees (3–5 °C) above the Tm of the primers used, while at the later cycles, it is a few degrees (3–5 °C) below the primer Tm. The higher temperatures give greater specificity for primer binding, and the lower temperatures permit more efficient amplification from the specific products formed during the initial cycles.[75]
- Universal Fast Walking: for genome walking and genetic fingerprinting using a more specific ‘two-sided’ PCR than conventional ‘one-sided’ approaches (using only one gene-specific primer and one general primer—which can lead to artefactual ‘noise’)[76] by virtue of a mechanism involving lariat structure formation. Streamlined derivatives of UFW are LaNe RAGE (lariat-dependent nested PCR for rapid amplification of genomic DNA ends),[77] 5’RACE LaNe[78] and 3’RACE LaNe.[79]
History[edit]

Diagrammatic representation of an example primer pair. The use of primers in an in vitro assay to allow DNA synthesis was a major innovation that allowed the development of PCR.
The heat-resistant enzymes that are a key component in polymerase chain reaction were discovered in the 1960s as a product of a microbial life form that lived in the superheated waters of Yellowstone’s Mushroom Spring.[80]
A 1971 paper in the Journal of Molecular Biology by Kjell Kleppe and co-workers in the laboratory of H. Gobind Khorana first described a method of using an enzymatic assay to replicate a short DNA template with primers in vitro.[81] However, this early manifestation of the basic PCR principle did not receive much attention at the time and the invention of the polymerase chain reaction in 1983 is generally credited to Kary Mullis.[82][page needed]

«Baby Blue», a 1986 prototype machine for doing PCR
When Mullis developed the PCR in 1983, he was working in Emeryville, California for Cetus Corporation, one of the first biotechnology companies, where he was responsible for synthesizing short chains of DNA. Mullis has written that he conceived the idea for PCR while cruising along the Pacific Coast Highway one night in his car.[83] He was playing in his mind with a new way of analyzing changes (mutations) in DNA when he realized that he had instead invented a method of amplifying any DNA region through repeated cycles of duplication driven by DNA polymerase. In Scientific American, Mullis summarized the procedure: «Beginning with a single molecule of the genetic material DNA, the PCR can generate 100 billion similar molecules in an afternoon. The reaction is easy to execute. It requires no more than a test tube, a few simple reagents, and a source of heat.»[84] DNA fingerprinting was first used for paternity testing in 1988.[85]
Mullis has credited his use of LSD as integral to his development of PCR: «Would I have invented PCR if I hadn’t taken LSD? I seriously doubt it. I could sit on a DNA molecule and watch the polymers go by. I learnt that partly on psychedelic drugs.»[86]
Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA,[1] were jointly awarded the Nobel Prize in Chemistry in 1993, seven years after Mullis and his colleagues at Cetus first put his proposal to practice.[87] Mullis’s 1985 paper with R. K. Saiki and H. A. Erlich, «Enzymatic Amplification of β-globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia»—the polymerase chain reaction invention (PCR)—was honored by a Citation for Chemical Breakthrough Award from the Division of History of Chemistry of the American Chemical Society in 2017.[88][2]
At the core of the PCR method is the use of a suitable DNA polymerase able to withstand the high temperatures of >90 °C (194 °F) required for separation of the two DNA strands in the DNA double helix after each replication cycle. The DNA polymerases initially employed for in vitro experiments presaging PCR were unable to withstand these high temperatures.[2] So the early procedures for DNA replication were very inefficient and time-consuming, and required large amounts of DNA polymerase and continuous handling throughout the process.
The discovery in 1976 of Taq polymerase—a DNA polymerase purified from the thermophilic bacterium, Thermus aquaticus, which naturally lives in hot (50 to 80 °C (122 to 176 °F)) environments[14] such as hot springs—paved the way for dramatic improvements of the PCR method. The DNA polymerase isolated from T. aquaticus is stable at high temperatures remaining active even after DNA denaturation,[15] thus obviating the need to add new DNA polymerase after each cycle.[3] This allowed an automated thermocycler-based process for DNA amplification.
Patent disputes[edit]
The PCR technique was patented by Kary Mullis and assigned to Cetus Corporation, where Mullis worked when he invented the technique in 1983. The Taq polymerase enzyme was also covered by patents. There have been several high-profile lawsuits related to the technique, including an unsuccessful lawsuit brought by DuPont. The Swiss pharmaceutical company Hoffmann-La Roche purchased the rights to the patents in 1992. The last of the commercial PCR patents expired in 2017.[89]
A related patent battle over the Taq polymerase enzyme is still ongoing[as of?] in several jurisdictions around the world between Roche and Promega. The legal arguments have extended beyond the lives of the original PCR and Taq polymerase patents, which expired on 28 March 2005.[90]
See also[edit]
- COVID-19 testing
- DNA spiking
- Loop-mediated isothermal amplification
- Selector technique
- Thermus thermophilus
- Pfu DNA polymerase
References[edit]
- ^ a b «The Nobel Prize in Chemistry 1993». NobelPrize.org.
- ^ a b c d Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N (December 1985). «Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia». Science. 230 (4732): 1350–54. Bibcode:1985Sci…230.1350S. doi:10.1126/science.2999980. PMID 2999980.
- ^ a b Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, et al. (January 1988). «Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase». Science. 239 (4839): 487–91. Bibcode:1988Sci…239..487S. doi:10.1126/science.239.4839.487. PMID 2448875.
- ^ Enners, Edward; Porta, Angela R. (2012). «Determining Annealing Temperatures for Polymerase Chain Reaction». The American Biology Teacher. 74 (4): 256–60. doi:10.1525/abt.2012.74.4.9. S2CID 86708426.
- ^ a b c d e f Ninfa, Alexander; Ballou, David; Benore, Marilee (2009). Fundamental Laboratory Approaches for Biochemistry and Biotechnology. United States: Wiley. pp. 408–10. ISBN 978-0-470-08766-4.
- ^ Cheng S, Fockler C, Barnes WM, Higuchi R (June 1994). «Effective amplification of long targets from cloned inserts and human genomic DNA». Proceedings of the National Academy of Sciences of the United States of America. 91 (12): 5695–99. Bibcode:1994PNAS…91.5695C. doi:10.1073/pnas.91.12.5695. PMC 44063. PMID 8202550.
- ^ Carr AC, Moore SD (2012). Lucia A (ed.). «Robust quantification of polymerase chain reactions using global fitting». PLOS ONE. 7 (5): e37640. Bibcode:2012PLoSO…737640C. doi:10.1371/journal.pone.0037640. PMC 3365123. PMID 22701526.
- ^ a b Joseph Sambrook & David W. Russel (2001). Molecular Cloning: A Laboratory Manual (third ed.). Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. ISBN 978-0-879-69576-7. Chapter 8: In vitro Amplification of DNA by the Polymerase Chain Reaction
- ^ «Polymerase Chain Reaction (PCR)». National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ «PCR». Genetic Science Learning Center, University of Utah.
- ^ Pavlov AR, Pavlova NV, Kozyavkin SA, Slesarev AI (May 2004). «Recent developments in the optimization of thermostable DNA polymerases for efficient applications». Trends in Biotechnology. 22 (5): 253–60. doi:10.1016/j.tibtech.2004.02.011. PMID 15109812.
- ^ Rychlik W, Spencer WJ, Rhoads RE (November 1990). «Optimization of the annealing temperature for DNA amplification in vitro». Nucleic Acids Research. 18 (21): 6409–12. doi:10.1093/nar/18.21.6409. PMC 332522. PMID 2243783.
- ^ a b Sharkey DJ, Scalice ER, Christy KG, Atwood SM, Daiss JL (May 1994). «Antibodies as thermolabile switches: high temperature triggering for the polymerase chain reaction». Bio/Technology. 12 (5): 506–09. doi:10.1038/nbt0594-506. PMID 7764710. S2CID 2885453.
- ^ a b Chien A, Edgar DB, Trela JM (September 1976). «Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus». Journal of Bacteriology. 127 (3): 1550–57. doi:10.1128/jb.127.3.1550-1557.1976. PMC 232952. PMID 8432.
- ^ a b Lawyer FC, Stoffel S, Saiki RK, Chang SY, Landre PA, Abramson RD, Gelfand DH (May 1993). «High-level expression, purification, and enzymatic characterization of full-length Thermus aquaticus DNA polymerase and a truncated form deficient in 5′ to 3′ exonuclease activity». PCR Methods and Applications. 2 (4): 275–87. doi:10.1101/gr.2.4.275. PMID 8324500.
- ^ a b c d Schochetman G, Ou CY, Jones WK (December 1988). «Polymerase chain reaction». The Journal of Infectious Diseases. 158 (6): 1154–57. doi:10.1093/infdis/158.6.1154. JSTOR 30137034. PMID 2461996.
- ^ Borman, Jon; Schuster, David; Li, Wu-bo; Jessee, Joel; Rashtchian, Ayoub (2000). «PCR from problematic templates» (PDF). Focus. 22 (1): 10. Archived from the original (PDF) on 7 March 2017.
- ^ Bogetto, Prachi; Waidne, Lisa; Anderson, Holly (2000). «Helpful tips for PCR» (PDF). Focus. 22 (1): 12. Archived from the original (PDF) on 7 March 2017.
- ^ Biolabs, New England. «Q5® High-Fidelity DNA Polymerase | NEB». www.neb.com. Retrieved 4 December 2021.
- ^ Sze, Marc A.; Schloss, Patrick D. (2019). «The Impact of DNA Polymerase and Number of Rounds of Amplification in PCR on 16S rRNA Gene Sequence Data». mSphere. 4 (3): e00163–19. doi:10.1128/mSphere.00163-19. PMC 6531881. PMID 31118299.
- ^ Sarkar G, Kapelner S, Sommer SS (December 1990). «Formamide can dramatically improve the specificity of PCR». Nucleic Acids Research. 18 (24): 7465. doi:10.1093/nar/18.24.7465. PMC 332902. PMID 2259646.
- ^ «Electronic PCR». NCBI – National Center for Biotechnology Information. Retrieved 13 March 2012.
- ^ Pavlov AR, Pavlova NV, Kozyavkin SA, Slesarev AI (2006). «Thermostable DNA Polymerases for a Wide Spectrum of Applications: Comparison of a Robust Hybrid TopoTaq to other enzymes». In Kieleczawa J (ed.). DNA Sequencing II: Optimizing Preparation and Cleanup. Jones & Bartlett. pp. 241–57. ISBN 978-0-7637-3383-4.
- ^ Pombert JF, Sistek V, Boissinot M, Frenette M (October 2009). «Evolutionary relationships among salivarius streptococci as inferred from multilocus phylogenies based on 16S rRNA-encoding, recA, secA, and secY gene sequences». BMC Microbiology. 9: 232. doi:10.1186/1471-2180-9-232. PMC 2777182. PMID 19878555.
- ^ «Chemical Synthesis, Sequencing, and Amplification of DNA (class notes on MBB/BIO 343)». Arizona State University. Archived from the original on 9 October 1997. Retrieved 29 October 2007.
- ^ Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. (April 2009). «The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments» (PDF). Clinical Chemistry. 55 (4): 611–22. doi:10.1373/clinchem.2008.112797. PMID 19246619.
- ^ a b c d Garibyan L, Avashia N (March 2013). «Polymerase chain reaction». The Journal of Investigative Dermatology. 133 (3): 1–4. doi:10.1038/jid.2013.1. PMC 4102308. PMID 23399825.
- ^ Schnell, S.; Mendoza, C. (October 1997). «Theoretical Description of the Polymerase Chain Reaction». Journal of Theoretical Biology. 188 (3): 313–18. Bibcode:1997JThBi.188..313S. doi:10.1006/jtbi.1997.0473. PMID 9344735.
- ^ Schnell, S.; Mendoza, C. (21 February 1997). «Enzymological Considerations for the Theoretical Description of the Quantitative Competitive Polymerase Chain Reaction (QC-PCR)». Journal of Theoretical Biology. 184 (4): 433–40. Bibcode:1997JThBi.184..433S. doi:10.1006/jtbi.1996.0283. ISSN 0022-5193. PMID 9082073.
- ^ Becker, Sven; Böger, Peter; Oehlmann, Ralfh; Ernst, Anneliese (1 November 2000). «PCR Bias in Ecological Analysis: a Case Study for Quantitative Taq Nuclease Assays in Analyses of Microbial Communities». Applied and Environmental Microbiology. 66 (11): 4945–53. Bibcode:2000ApEnM..66.4945B. doi:10.1128/AEM.66.11.4945-4953.2000. ISSN 1098-5336. PMC 92404. PMID 11055948.
- ^ Solomon, Anthony W.; Peeling, Rosanna W.; Foster, Allen; Mabey, David C. W. (1 October 2004). «Diagnosis and Assessment of Trachoma». Clinical Microbiology Reviews. 17 (4): 982–1011. doi:10.1128/CMR.17.4.982-1011.2004. ISSN 0893-8512. PMC 523557. PMID 15489358.
- ^ Ramzy, Reda M.R. (April 2002). «Recent advances in molecular diagnostic techniques for human lymphatic filariasis and their use in epidemiological research». Transactions of the Royal Society of Tropical Medicine and Hygiene. 96: S225–29. doi:10.1016/S0035-9203(02)90080-5. PMID 12055843.
- ^ Sachse, Konrad (2003). «Specificity and performance of diagnostic PCR assays». In Sachse, Konrad; Frey, Joachim (eds.). PCR Detection of Microbial Pathogens. Methods in Molecular Biology. Vol. 216. Totowa, New Jersey: Humana Press. pp. 3–29. doi:10.1385/1-59259-344-5:03. ISBN 978-1-59259-344-6. PMID 12512353.
- ^ Quill E (March 2008). «Medicine. Blood-matching goes genetic». Science. 319 (5869): 1478–79. doi:10.1126/science.319.5869.1478. PMID 18339916. S2CID 36945291.
- ^ Tomar, Rukam (2010). Molecular Markers and Plant Biotechnology. Pitman Pura, New Delhi: New India Publishing Agency. p. 188. ISBN 978-93-80235-25-7.
- ^ a b c Cai HY, Caswell JL, Prescott JF (March 2014). «Nonculture molecular techniques for diagnosis of bacterial disease in animals: a diagnostic laboratory perspective». Veterinary Pathology. 51 (2): 341–50. doi:10.1177/0300985813511132. PMID 24569613.
- ^ Salis AD (2009). «Applications in Clinical Microbiology». Real-Time PCR: Current Technology and Applications. Caister Academic Press. ISBN 978-1-904455-39-4.
- ^ Kwok S, Mack DH, Mullis KB, Poiesz B, Ehrlich G, Blair D, et al. (May 1987). «Identification of human immunodeficiency virus sequences by using in vitro enzymatic amplification and oligomer cleavage detection». Journal of Virology. 61 (5): 1690–94. doi:10.1128/jvi.61.5.1690-1694.1987. PMC 254157. PMID 2437321.
- ^ «Coronavirus: il viaggio dei test». Istituto Superiore di Sanità.
- ^ Finger, Horst; von Koenig, Carl Heinz Wirsing (1996). Baron, Samuel (ed.). Medical Microbiology (4th ed.). Galveston, TX: University of Texas Medical Branch at Galveston. ISBN 978-0-9631172-1-2. PMID 21413270.
- ^ Yeh, Sylvia H.; Mink, ChrisAnna M. (2012). «Bordetella pertussis and Pertussis (Whooping Cough)». Netter’s Infectious Diseases. Netter’s Infectious Diseases. pp. 11–14. doi:10.1016/B978-1-4377-0126-5.00003-3. ISBN 978-1-4377-0126-5.
- ^ Alonso A, Martín P, Albarrán C, García P, García O, de Simón LF, et al. (January 2004). «Real-Time PCR Designs to Estimate Nuclear and Mitochondrial DNA Copy Number in Forensic and Ancient DNA Studies». Forensic Science International. 139 (2–3): 141–49. doi:10.1016/j.forsciint.2003.10.008. PMID 15040907.
- ^ Boehnke M, Arnheim N, Li H, Collins FS (July 1989). «Fine-structure genetic mapping of human chromosomes using the polymerase chain reaction on single sperm: experimental design considerations». American Journal of Human Genetics. 45 (1): 21–32. PMC 1683385. PMID 2568090.
- ^ Zhou YH, Zhang XP, Ebright RH (November 1991). «Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase». Nucleic Acids Research. 19 (21): 6052. doi:10.1093/nar/19.21.6052. PMC 329070. PMID 1658751.
- ^ Stursberg, Stephanie (2021). «Setting up a PCR lab from scratch». INTEGRA Biosciences.
{{cite web}}: CS1 maint: url-status (link) - ^ Bulduk, BK, et al. (March 2020). «A Novel Amplification-Refractory Mutation System-PCR Strategy to Screen MT-TL1 Pathogenic Variants in Patient Repositories». Genet Test Mol Biomarkers. 24 (3): 165–170. doi:10.1089/gtmb.2019.0079. PMID 32167396.
- ^ Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, et al. (April 1989). «Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS)». Nucleic Acids Research. 17 (7): 2503–16. doi:10.1093/nar/17.7.2503. PMC 317639. PMID 2785681.
- ^ Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL (October 1995). «Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides». Gene. 164 (1): 49–53. doi:10.1016/0378-1119(95)00511-4. PMID 7590320.
- ^ Innis MA, Myambo KB, Gelfand DH, Brow MA (December 1988). «DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA». Proceedings of the National Academy of Sciences of the United States of America. 85 (24): 9436–40. Bibcode:1988PNAS…85.9436I. doi:10.1073/pnas.85.24.9436. PMC 282767. PMID 3200828.
- ^ Pierce KE, Wangh LJ (2007). Linear-after-the-exponential polymerase chain reaction and allied technologies Real-time detection strategies for rapid, reliable diagnosis from single cells. Methods in Molecular Medicine. Vol. 132. pp. 65–85. doi:10.1007/978-1-59745-298-4_7. ISBN 978-1-58829-578-1. PMID 17876077.
- ^ Krishnan M, Ugaz VM, Burns MA (October 2002). «PCR in a Rayleigh-Bénard convection cell». Science. 298 (5594): 793. doi:10.1126/science.298.5594.793. PMID 12399582.
- ^ Priye A, Hassan YA, Ugaz VM (November 2013). «Microscale chaotic advection enables robust convective DNA replication». Analytical Chemistry. 85 (21): 10536–41. doi:10.1021/ac402611s. PMID 24083802.
- ^ Schwartz JJ, Lee C, Shendure J (September 2012). «Accurate gene synthesis with tag-directed retrieval of sequence-verified DNA molecules». Nature Methods. 9 (9): 913–15. doi:10.1038/nmeth.2137. PMC 3433648. PMID 22886093.
- ^ Vincent M, Xu Y, Kong H (August 2004). «Helicase-dependent isothermal DNA amplification». EMBO Reports. 5 (8): 795–800. doi:10.1038/sj.embor.7400200. PMC 1249482. PMID 15247927.
- ^ Chou Q, Russell M, Birch DE, Raymond J, Bloch W (April 1992). «Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications». Nucleic Acids Research. 20 (7): 1717–23. doi:10.1093/nar/20.7.1717. PMC 312262. PMID 1579465.
- ^ Kellogg DE, Rybalkin I, Chen S, Mukhamedova N, Vlasik T, Siebert PD, Chenchik A (June 1994). «TaqStart Antibody: «hot start» PCR facilitated by a neutralizing monoclonal antibody directed against Taq DNA polymerase». BioTechniques. 16 (6): 1134–37. PMID 8074881.
- ^ San Millán RM, Martínez-Ballesteros I, Rementeria A, Garaizar J, Bikandi J (December 2013). «Online exercise for the design and simulation of PCR and PCR-RFLP experiments». BMC Research Notes. 6: 513. doi:10.1186/1756-0500-6-513. PMC 4029544. PMID 24314313.
- ^ Zietkiewicz E, Rafalski A, Labuda D (March 1994). «Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification». Genomics. 20 (2): 176–83. doi:10.1006/geno.1994.1151. PMID 8020964.
- ^ Ochman H, Gerber AS, Hartl DL (November 1988). «Genetic applications of an inverse polymerase chain reaction». Genetics. 120 (3): 621–23. doi:10.1093/genetics/120.3.621. PMC 1203539. PMID 2852134.
- ^ Mueller PR, Wold B (November 1989). «In vivo footprinting of a muscle specific enhancer by ligation mediated PCR». Science. 246 (4931): 780–86. Bibcode:1989Sci…246..780M. doi:10.1126/science.2814500. PMID 2814500.
- ^ Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (September 1996). «Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands». Proceedings of the National Academy of Sciences of the United States of America. 93 (18): 9821–26. Bibcode:1996PNAS…93.9821H. doi:10.1073/pnas.93.18.9821. PMC 38513. PMID 8790415.
- ^ Isenbarger TA, Finney M, Ríos-Velázquez C, Handelsman J, Ruvkun G (February 2008). «Miniprimer PCR, a new lens for viewing the microbial world». Applied and Environmental Microbiology. 74 (3): 840–49. Bibcode:2008ApEnM..74..840I. doi:10.1128/AEM.01933-07. PMC 2227730. PMID 18083877.
- ^ Shen C, Yang W, Ji Q, Maki H, Dong A, Zhang Z (November 2009). «NanoPCR observation: different levels of DNA replication fidelity in nanoparticle-enhanced polymerase chain reactions». Nanotechnology. 20 (45): 455103. Bibcode:2009Nanot..20S5103S. doi:10.1088/0957-4484/20/45/455103. PMID 19822925. S2CID 3393115.
- ^ Shen, Cenchao (2013). «An Overview of Nanoparticle-Assisted Polymerase Chain Reaction Technology». An Overview of Nanoparticle‐Assisted Polymerase Chain Reaction Technology. US: Wiley-Blackwell Publishing Ltd. pp. 97–106. doi:10.1002/9781118451915.ch5. ISBN 978-1-118-45191-5.
- ^ Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (April 1989). «Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension». Gene. 77 (1): 61–68. doi:10.1016/0378-1119(89)90359-4. PMID 2744488.
- ^ Moller, Simon (2006). PCR: The Basics. US: Taylor & Francis Group. p. 144. ISBN 978-0-415-35547-6.
- ^ David F, Turlotte E (November 1998). «[A method of isothermal gene amplification]» [An Isothermal Amplification Method]. Comptes Rendus de l’Académie des Sciences, Série III. 321 (11): 909–14. Bibcode:1998CRASG.321..909D. doi:10.1016/S0764-4469(99)80005-5. PMID 9879470.
- ^ Fabrice David (September–October 2002). «Utiliser les propriétés topologiques de l’ADN: une nouvelle arme contre les agents pathogènes» (PDF). Fusion. Archived from the original (PDF) on 28 November 2007.(in French)
- ^ Dobosy JR, Rose SD, Beltz KR, Rupp SM, Powers KM, Behlke MA, Walder JA (August 2011). «RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers». BMC Biotechnology. 11: 80. doi:10.1186/1472-6750-11-80. PMC 3224242. PMID 21831278.
- ^ Shyamala, V.; Ferro-Luzzi, Ames G. (1993). Single Specific Primer-Polymerase Chain Reaction (SSP-PCR) and Genome Walking. Methods in Molecular Biology. Vol. 15. pp. 339–48. doi:10.1385/0-89603-244-2:339. ISBN 978-0-89603-244-6. PMID 21400290.
- ^ Bing DH, Boles C, Rehman FN, Audeh M, Belmarsh M, Kelley B, Adams CP (1996). «Bridge amplification: a solid phase PCR system for the amplification and detection of allelic differences in single copy genes». Genetic Identity Conference Proceedings, Seventh International Symposium on Human Identification. Archived from the original on 7 May 2001.
- ^ Khan Z, Poetter K, Park DJ (April 2008). «Enhanced solid phase PCR: mechanisms to increase priming by solid support primers». Analytical Biochemistry. 375 (2): 391–93. doi:10.1016/j.ab.2008.01.021. PMID 18267099.
- ^ Raoult D, Aboudharam G, Crubézy E, Larrouy G, Ludes B, Drancourt M (November 2000). «Molecular identification by «suicide PCR» of Yersinia pestis as the agent of medieval black death». Proceedings of the National Academy of Sciences of the United States of America. 97 (23): 12800–03. Bibcode:2000PNAS…9712800R. doi:10.1073/pnas.220225197. PMC 18844. PMID 11058154.
- ^ Liu YG, Whittier RF (February 1995). «Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking». Genomics. 25 (3): 674–81. doi:10.1016/0888-7543(95)80010-J. PMID 7759102.
- ^ Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (July 1991). «‘Touchdown’ PCR to circumvent spurious priming during gene amplification». Nucleic Acids Research. 19 (14): 4008. doi:10.1093/nar/19.14.4008. PMC 328507. PMID 1861999.
- ^ Myrick KV, Gelbart WM (February 2002). «Universal Fast Walking for direct and versatile determination of flanking sequence». Gene. 284 (1–2): 125–31. doi:10.1016/S0378-1119(02)00384-0. PMID 11891053.
- ^ «Full Text – LaNe RAGE: a new tool for genomic DNA flanking sequence determination». www.ejbiotechnology.info.
- ^ Park DJ (January 2005). «A new 5′ terminal murine GAPDH exon identified using 5’RACE LaNe». Molecular Biotechnology. 29 (1): 39–46. doi:10.1385/MB:29:1:39. PMID 15668518. S2CID 45702164.
- ^ Park DJ (April 2004). «3′ RACE LaNe: a simple and rapid fully nested PCR method to determine 3′-terminal cDNA sequence». BioTechniques. 36 (4): 586–88, 590. doi:10.2144/04364BM04. PMID 15088375.
- ^ «Key ingredient in coronavirus tests comes from Yellowstone’s lakes». Science. 31 March 2020. Retrieved 13 May 2020.
- ^ Kleppe K, Ohtsuka E, Kleppe R, Molineux I, Khorana HG (March 1971). «Studies on polynucleotides. XCVI. Repair replications of short synthetic DNA’s as catalyzed by DNA polymerases». Journal of Molecular Biology. 56 (2): 341–61. doi:10.1016/0022-2836(71)90469-4. PMID 4927950.
- ^ Rabinow, Paul (1996). Making PCR: A Story of Biotechnology. Chicago: University of Chicago Press. ISBN 978-0-226-70146-2.
- ^ Mullis, Kary (1998). Dancing Naked in the Mind Field. New York: Pantheon Books. ISBN 978-0-679-44255-4.
- ^ Mullis KB (April 1990). «The unusual origin of the polymerase chain reaction». Scientific American. 262 (4): 56–61, 64–65. Bibcode:1990SciAm.262d..56M. doi:10.1038/scientificamerican0490-56. PMID 2315679.
- ^ Patidar M, Agrawal S, Parveen F, Khare P (2015). «Molecular insights of saliva in solving paternity dispute». Journal of Forensic Dental Sciences. 7 (1): 76–79. doi:10.4103/0975-1475.150325. PMC 4330625. PMID 25709326.
- ^ Nichols D, Barker E (2016). «Psychedelics». Pharmacological Reviews. 68 (2): 264–355. doi:10.1124/pr.115.011478. PMC 4813425. PMID 26841800.
- ^ «The Nobel Prize in Chemistry 1993». NobelPrize.org.
- ^ «Citations for Chemical Breakthrough Awards 2017 Awardees». Division of the History of Chemistry. Retrieved 12 March 2018.
- ^ Fore, J. Jr.; Wiechers, I. R.; Cook-Deegan, R. (3 July 2006). «The effects of business practices, licensing, and intellectual property on development and dissemination of the polymerase chain reaction: case study». Journal of Biomedical Discovery and Collaboration. 1: 7. doi:10.1186/1747-5333-1-7. PMC 1523369. PMID 16817955.
- ^ «Advice on How to Survive the Taq Wars». GEN Genetic Engineering News – Biobusiness Channel. 26 (9). 1 May 2006.
External links[edit]
- US Patent for PCR
- What is PCR plateau effect? YouTube tutorial video
- History of the Polymerase Chain Reaction from the Smithsonian Institution Archives
A strip of eight PCR tubes, each containing a 100 μL reaction mixture
The polymerase chain reaction (PCR) is a method widely used to rapidly make millions to billions of copies (complete or partial) of a specific DNA sample, allowing scientists to take a very small sample of DNA and amplify it (or a part of it) to a large enough amount to study in detail. PCR was invented in 1983 by the American biochemist Kary Mullis at Cetus Corporation; Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA,[1] were jointly awarded the Nobel Prize in Chemistry in 1993.
PCR is fundamental to many of the procedures used in genetic testing and research, including analysis of ancient samples of DNA and identification of infectious agents. Using PCR, copies of very small amounts of DNA sequences are exponentially amplified in a series of cycles of temperature changes. PCR is now a common and often indispensable technique used in medical laboratory research for a broad variety of applications including biomedical research and criminal forensics.[2][3]
The majority of PCR methods rely on thermal cycling. Thermal cycling exposes reactants to repeated cycles of heating and cooling to permit different temperature-dependent reactions—specifically, DNA melting and enzyme-driven DNA replication. PCR employs two main reagents—primers (which are short single strand DNA fragments known as oligonucleotides that are a complementary sequence to the target DNA region) and a DNA polymerase. In the first step of PCR, the two strands of the DNA double helix are physically separated at a high temperature in a process called nucleic acid denaturation. In the second step, the temperature is lowered and the primers bind to the complementary sequences of DNA. The two DNA strands then become templates for DNA polymerase to enzymatically assemble a new DNA strand from free nucleotides, the building blocks of DNA. As PCR progresses, the DNA generated is itself used as a template for replication, setting in motion a chain reaction in which the original DNA template is exponentially amplified.
Almost all PCR applications employ a heat-stable DNA polymerase, such as Taq polymerase, an enzyme originally isolated from the thermophilic bacterium Thermus aquaticus. If the polymerase used was heat-susceptible, it would denature under the high temperatures of the denaturation step. Before the use of Taq polymerase, DNA polymerase had to be manually added every cycle, which was a tedious and costly process.[4]
Applications of the technique include DNA cloning for sequencing, gene cloning and manipulation, gene mutagenesis; construction of DNA-based phylogenies, or functional analysis of genes; diagnosis and monitoring of genetic disorders; amplification of ancient DNA;[5] analysis of genetic fingerprints for DNA profiling (for example, in forensic science and parentage testing); and detection of pathogens in nucleic acid tests for the diagnosis of infectious diseases.
Principles[edit]
PCR amplifies a specific region of a DNA strand (the DNA target). Most PCR methods amplify DNA fragments of between 0.1 and 10 kilo base pairs (kbp) in length, although some techniques allow for amplification of fragments up to 40 kbp.[6] The amount of amplified product is determined by the available substrates in the reaction, which becomes limiting as the reaction progresses.[7]
A basic PCR set-up requires several components and reagents,[8] including:
- a DNA template that contains the DNA target region to amplify
- a DNA polymerase; an enzyme that polymerizes new DNA strands; heat-resistant Taq polymerase is especially common,[9] as it is more likely to remain intact during the high-temperature DNA denaturation process
- two DNA primers that are complementary to the 3′ (three prime) ends of each of the sense and anti-sense strands of the DNA target (DNA polymerase can only bind to and elongate from a double-stranded region of DNA; without primers, there is no double-stranded initiation site at which the polymerase can bind);[10] specific primers that are complementary to the DNA target region are selected beforehand, and are often custom-made in a laboratory or purchased from commercial biochemical suppliers
- deoxynucleoside triphosphates, or dNTPs (sometimes called «deoxynucleotide triphosphates»; nucleotides containing triphosphate groups), the building blocks from which the DNA polymerase synthesizes a new DNA strand
- a buffer solution providing a suitable chemical environment for optimum activity and stability of the DNA polymerase
- bivalent cations, typically magnesium (Mg) or manganese (Mn) ions; Mg2+ is the most common, but Mn2+ can be used for PCR-mediated DNA mutagenesis, as a higher Mn2+ concentration increases the error rate during DNA synthesis;[11] and monovalent cations, typically potassium (K) ions[better source needed]
The reaction is commonly carried out in a volume of 10–200 μL in small reaction tubes (0.2–0.5 mL volumes) in a thermal cycler. The thermal cycler heats and cools the reaction tubes to achieve the temperatures required at each step of the reaction (see below). Many modern thermal cyclers make use of the Peltier effect, which permits both heating and cooling of the block holding the PCR tubes simply by reversing the electric current. Thin-walled reaction tubes permit favorable thermal conductivity to allow for rapid thermal equilibrium. Most thermal cyclers have heated lids to prevent condensation at the top of the reaction tube. Older thermal cyclers lacking a heated lid require a layer of oil on top of the reaction mixture or a ball of wax inside the tube.
Procedure[edit]
Typically, PCR consists of a series of 20–40 repeated temperature changes, called thermal cycles, with each cycle commonly consisting of two or three discrete temperature steps (see figure below). The cycling is often preceded by a single temperature step at a very high temperature (>90 °C (194 °F)), and followed by one hold at the end for final product extension or brief storage. The temperatures used and the length of time they are applied in each cycle depend on a variety of parameters, including the enzyme used for DNA synthesis, the concentration of bivalent ions and dNTPs in the reaction, and the melting temperature (Tm) of the primers.[12] The individual steps common to most PCR methods are as follows:
- Initialization: This step is only required for DNA polymerases that require heat activation by hot-start PCR.[13] It consists of heating the reaction chamber to a temperature of 94–96 °C (201–205 °F), or 98 °C (208 °F) if extremely thermostable polymerases are used, which is then held for 1–10 minutes.
- Denaturation: This step is the first regular cycling event and consists of heating the reaction chamber to 94–98 °C (201–208 °F) for 20–30 seconds. This causes DNA melting, or denaturation, of the double-stranded DNA template by breaking the hydrogen bonds between complementary bases, yielding two single-stranded DNA molecules.
- Annealing: In the next step, the reaction temperature is lowered to 50–65 °C (122–149 °F) for 20–40 seconds, allowing annealing of the primers to each of the single-stranded DNA templates. Two different primers are typically included in the reaction mixture: one for each of the two single-stranded complements containing the target region. The primers are single-stranded sequences themselves, but are much shorter than the length of the target region, complementing only very short sequences at the 3′ end of each strand.
- It is critical to determine a proper temperature for the annealing step because efficiency and specificity are strongly affected by the annealing temperature. This temperature must be low enough to allow for hybridization of the primer to the strand, but high enough for the hybridization to be specific, i.e., the primer should bind only to a perfectly complementary part of the strand, and nowhere else. If the temperature is too low, the primer may bind imperfectly. If it is too high, the primer may not bind at all. A typical annealing temperature is about 3–5 °C below the Tm of the primers used. Stable hydrogen bonds between complementary bases are formed only when the primer sequence very closely matches the template sequence. During this step, the polymerase binds to the primer-template hybrid and begins DNA formation.
- Extension/elongation: The temperature at this step depends on the DNA polymerase used; the optimum activity temperature for the thermostable DNA polymerase of Taq polymerase is approximately 75–80 °C (167–176 °F),[14][15] though a temperature of 72 °C (162 °F) is commonly used with this enzyme. In this step, the DNA polymerase synthesizes a new DNA strand complementary to the DNA template strand by adding free dNTPs from the reaction mixture that is complementary to the template in the 5′-to-3′ direction, condensing the 5′-phosphate group of the dNTPs with the 3′-hydroxy group at the end of the nascent (elongating) DNA strand. The precise time required for elongation depends both on the DNA polymerase used and on the length of the DNA target region to amplify. As a rule of thumb, at their optimal temperature, most DNA polymerases polymerize a thousand bases per minute. Under optimal conditions (i.e., if there are no limitations due to limiting substrates or reagents), at each extension/elongation step, the number of DNA target sequences is doubled. With each successive cycle, the original template strands plus all newly generated strands become template strands for the next round of elongation, leading to exponential (geometric) amplification of the specific DNA target region.
- The processes of denaturation, annealing and elongation constitute a single cycle. Multiple cycles are required to amplify the DNA target to millions of copies. The formula used to calculate the number of DNA copies formed after a given number of cycles is 2n, where n is the number of cycles. Thus, a reaction set for 30 cycles results in 230, or 1,073,741,824, copies of the original double-stranded DNA target region.
- Final elongation: This single step is optional, but is performed at a temperature of 70–74 °C (158–165 °F) (the temperature range required for optimal activity of most polymerases used in PCR) for 5–15 minutes after the last PCR cycle to ensure that any remaining single-stranded DNA is fully elongated.
- Final hold: The final step cools the reaction chamber to 4–15 °C (39–59 °F) for an indefinite time, and may be employed for short-term storage of the PCR products.
Ethidium bromide-stained PCR products after gel electrophoresis. Two sets of primers were used to amplify a target sequence from three different tissue samples. No amplification is present in sample #1; DNA bands in sample #2 and #3 indicate successful amplification of the target sequence. The gel also shows a positive control, and a DNA ladder containing DNA fragments of defined length for sizing the bands in the experimental PCRs.
To check whether the PCR successfully generated the anticipated DNA target region (also sometimes referred to as the amplimer or amplicon), agarose gel electrophoresis may be employed for size separation of the PCR products. The size of the PCR products is determined by comparison with a DNA ladder, a molecular weight marker which contains DNA fragments of known sizes, which runs on the gel alongside the PCR products.
Stages[edit]
Exponential amplification
As with other chemical reactions, the reaction rate and efficiency of PCR are affected by limiting factors. Thus, the entire PCR process can further be divided into three stages based on reaction progress:
- Exponential amplification: At every cycle, the amount of product is doubled (assuming 100% reaction efficiency). After 30 cycles, a single copy of DNA can be increased up to 1,000,000,000 (one billion) copies. In a sense, then, the replication of a discrete strand of DNA is being manipulated in a tube under controlled conditions.[16] The reaction is very sensitive: only minute quantities of DNA must be present.
- Leveling off stage: The reaction slows as the DNA polymerase loses activity and as consumption of reagents, such as dNTPs and primers, causes them to become more limited.
- Plateau: No more product accumulates due to exhaustion of reagents and enzyme.
Optimization[edit]
In practice, PCR can fail for various reasons, such as sensitivity or contamination.[17][18] Contamination with extraneous DNA can lead to spurious products and is addressed with lab protocols and procedures that separate pre-PCR mixtures from potential DNA contaminants.[8] For instance, if DNA from a crime scene is analyzed, a single DNA molecule from lab personnel could be amplified and misguide the investigation. Hence the PCR-setup areas is separated from the analysis or purification of other PCR products, disposable plasticware used, and the work surface between reaction setups needs to be thoroughly cleaned.
Specificity can be adjusted by experimental conditions so that no spurious products are generated. Primer-design techniques are important in improving PCR product yield and in avoiding the formation of unspecific products. The usage of alternate buffer components or polymerase enzymes can help with amplification of long or otherwise problematic regions of DNA. For instance, Q5 polymerase is said to be ~280 times less error-prone than Taq polymerase.[19][20] Both the running parameters (e.g. temperature and duration of cycles), or the addition of reagents, such as formamide, may increase the specificity and yield of PCR.[21] Computer simulations of theoretical PCR results (Electronic PCR) may be performed to assist in primer design.[22]
Applications[edit]
Selective DNA isolation[edit]
PCR allows isolation of DNA fragments from genomic DNA by selective amplification of a specific region of DNA. This use of PCR augments many ways, such as generating hybridization probes for Southern or northern hybridization and DNA cloning, which require larger amounts of DNA, representing a specific DNA region. PCR supplies these techniques with high amounts of pure DNA, enabling analysis of DNA samples even from very small amounts of starting material.
Other applications of PCR include DNA sequencing to determine unknown PCR-amplified sequences in which one of the amplification primers may be used in Sanger sequencing, isolation of a DNA sequence to expedite recombinant DNA technologies involving the insertion of a DNA sequence into a plasmid, phage, or cosmid (depending on size) or the genetic material of another organism. Bacterial colonies (such as E. coli) can be rapidly screened by PCR for correct DNA vector constructs.[23] PCR may also be used for genetic fingerprinting; a forensic technique used to identify a person or organism by comparing experimental DNAs through different PCR-based methods.
Electrophoresis of PCR-amplified DNA fragments:
- Father
- Child
- Mother
The child has inherited some, but not all, of the fingerprints of each of its parents, giving it a new, unique fingerprint.
Some PCR fingerprint methods have high discriminative power and can be used to identify genetic relationships between individuals, such as parent-child or between siblings, and are used in paternity testing (Fig. 4). This technique may also be used to determine evolutionary relationships among organisms when certain molecular clocks are used (i.e. the 16S rRNA and recA genes of microorganisms).[24]
Amplification and quantification of DNA[edit]
Because PCR amplifies the regions of DNA that it targets, PCR can be used to analyze extremely small amounts of sample. This is often critical for forensic analysis, when only a trace amount of DNA is available as evidence. PCR may also be used in the analysis of ancient DNA that is tens of thousands of years old. These PCR-based techniques have been successfully used on animals, such as a forty-thousand-year-old mammoth, and also on human DNA, in applications ranging from the analysis of Egyptian mummies to the identification of a Russian tsar and the body of English king Richard III.[25]
Quantitative PCR or Real Time PCR (qPCR,[26] not to be confused with RT-PCR) methods allow the estimation of the amount of a given sequence present in a sample—a technique often applied to quantitatively determine levels of gene expression. Quantitative PCR is an established tool for DNA quantification that measures the accumulation of DNA product after each round of PCR amplification.
qPCR allows the quantification and detection of a specific DNA sequence in real time since it measures concentration while the synthesis process is taking place. There are two methods for simultaneous detection and quantification. The first method consists of using fluorescent dyes that are retained nonspecifically in between the double strands. The second method involves probes that code for specific sequences and are fluorescently labeled. Detection of DNA using these methods can only be seen after the hybridization of probes with its complementary DNA (cDNA) takes place. An interesting technique combination is real-time PCR and reverse transcription. This sophisticated technique, called RT-qPCR, allows for the quantification of a small quantity of RNA. Through this combined technique, mRNA is converted to cDNA, which is further quantified using qPCR. This technique lowers the possibility of error at the end point of PCR,[27] increasing chances for detection of genes associated with genetic diseases such as cancer.[5] Laboratories use RT-qPCR for the purpose of sensitively measuring gene regulation. The mathematical foundations for the reliable quantification of the PCR[28] and RT-qPCR[29] facilitate the implementation of accurate fitting procedures of experimental data in research, medical, diagnostic and infectious disease applications.[30][31][32][33]
Medical and diagnostic applications[edit]
Prospective parents can be tested for being genetic carriers, or their children might be tested for actually being affected by a disease.[2] DNA samples for prenatal testing can be obtained by amniocentesis, chorionic villus sampling, or even by the analysis of rare fetal cells circulating in the mother’s bloodstream. PCR analysis is also essential to preimplantation genetic diagnosis, where individual cells of a developing embryo are tested for mutations.
- PCR can also be used as part of a sensitive test for tissue typing, vital to organ transplantation. As of 2008, there is even a proposal to replace the traditional antibody-based tests for blood type with PCR-based tests.[34]
- Many forms of cancer involve alterations to oncogenes. By using PCR-based tests to study these mutations, therapy regimens can sometimes be individually customized to a patient. PCR permits early diagnosis of malignant diseases such as leukemia and lymphomas, which is currently the highest-developed in cancer research and is already being used routinely. PCR assays can be performed directly on genomic DNA samples to detect translocation-specific malignant cells at a sensitivity that is at least 10,000 fold higher than that of other methods.[35] PCR is very useful in the medical field since it allows for the isolation and amplification of tumor suppressors. Quantitative PCR for example, can be used to quantify and analyze single cells, as well as recognize DNA, mRNA and protein confirmations and combinations.[27]
Infectious disease applications[edit]
PCR allows for rapid and highly specific diagnosis of infectious diseases, including those caused by bacteria or viruses.[36] PCR also permits identification of non-cultivatable or slow-growing microorganisms such as mycobacteria, anaerobic bacteria, or viruses from tissue culture assays and animal models. The basis for PCR diagnostic applications in microbiology is the detection of infectious agents and the discrimination of non-pathogenic from pathogenic strains by virtue of specific genes.[36][37]
Characterization and detection of infectious disease organisms have been revolutionized by PCR in the following ways:
- The human immunodeficiency virus (or HIV), is a difficult target to find and eradicate. The earliest tests for infection relied on the presence of antibodies to the virus circulating in the bloodstream. However, antibodies don’t appear until many weeks after infection, maternal antibodies mask the infection of a newborn, and therapeutic agents to fight the infection don’t affect the antibodies. PCR tests have been developed that can detect as little as one viral genome among the DNA of over 50,000 host cells.[38] Infections can be detected earlier, donated blood can be screened directly for the virus, newborns can be immediately tested for infection, and the effects of antiviral treatments can be quantified.
- Some disease organisms, such as that for tuberculosis, are difficult to sample from patients and slow to be grown in the laboratory. PCR-based tests have allowed detection of small numbers of disease organisms (both live or dead), in convenient samples. Detailed genetic analysis can also be used to detect antibiotic resistance, allowing immediate and effective therapy. The effects of therapy can also be immediately evaluated.
- The spread of a disease organism through populations of domestic or wild animals can be monitored by PCR testing. In many cases, the appearance of new virulent sub-types can be detected and monitored. The sub-types of an organism that were responsible for earlier epidemics can also be determined by PCR analysis.
- Viral DNA can be detected by PCR. The primers used must be specific to the targeted sequences in the DNA of a virus, and PCR can be used for diagnostic analyses or DNA sequencing of the viral genome. The high sensitivity of PCR permits virus detection soon after infection and even before the onset of disease.[36] Such early detection may give physicians a significant lead time in treatment. The amount of virus («viral load») in a patient can also be quantified by PCR-based DNA quantitation techniques (see below). A variant of PCR (RT-PCR) is used for detecting viral RNA rather than DNA: in this test the enzyme reverse transcriptase is used to generate a DNA sequence which matches the viral RNA; this DNA is then amplified as per the usual PCR method. RT-PCR is widely used to detect the SARS-CoV-2 viral genome.[39]
- Diseases such as pertussis (or whooping cough) are caused by the bacteria Bordetella pertussis. This bacteria is marked by a serious acute respiratory infection that affects various animals and humans and has led to the deaths of many young children. The pertussis toxin is a protein exotoxin that binds to cell receptors by two dimers and reacts with different cell types such as T lymphocytes which play a role in cell immunity.[40] PCR is an important testing tool that can detect sequences within the gene for the pertussis toxin. Because PCR has a high sensitivity for the toxin and a rapid turnaround time, it is very efficient for diagnosing pertussis when compared to culture.[41]
Forensic applications[edit]
The development of PCR-based genetic (or DNA) fingerprinting protocols has seen widespread application in forensics:
-
DNA samples are often taken at crime scenes and analyzed by PCR.
In its most discriminating form, genetic fingerprinting can uniquely discriminate any one person from the entire population of the world. Minute samples of DNA can be isolated from a crime scene, and compared to that from suspects, or from a DNA database of earlier evidence or convicts. Simpler versions of these tests are often used to rapidly rule out suspects during a criminal investigation. Evidence from decades-old crimes can be tested, confirming or exonerating the people originally convicted.
- Forensic DNA typing has been an effective way of identifying or exonerating criminal suspects due to analysis of evidence discovered at a crime scene. The human genome has many repetitive regions that can be found within gene sequences or in non-coding regions of the genome. Specifically, up to 40% of human DNA is repetitive.[5] There are two distinct categories for these repetitive, non-coding regions in the genome. The first category is called variable number tandem repeats (VNTR), which are 10–100 base pairs long and the second category is called short tandem repeats (STR) and these consist of repeated 2–10 base pair sections. PCR is used to amplify several well-known VNTRs and STRs using primers that flank each of the repetitive regions. The sizes of the fragments obtained from any individual for each of the STRs will indicate which alleles are present. By analyzing several STRs for an individual, a set of alleles for each person will be found that statistically is likely to be unique.[5] Researchers have identified the complete sequence of the human genome. This sequence can be easily accessed through the NCBI website and is used in many real-life applications. For example, the FBI has compiled a set of DNA marker sites used for identification, and these are called the Combined DNA Index System (CODIS) DNA database.[5] Using this database enables statistical analysis to be used to determine the probability that a DNA sample will match. PCR is a very powerful and significant analytical tool to use for forensic DNA typing because researchers only need a very small amount of the target DNA to be used for analysis. For example, a single human hair with attached hair follicle has enough DNA to conduct the analysis. Similarly, a few sperm, skin samples from under the fingernails, or a small amount of blood can provide enough DNA for conclusive analysis.[5]
- Less discriminating forms of DNA fingerprinting can help in DNA paternity testing, where an individual is matched with their close relatives. DNA from unidentified human remains can be tested, and compared with that from possible parents, siblings, or children. Similar testing can be used to confirm the biological parents of an adopted (or kidnapped) child. The actual biological father of a newborn can also be confirmed (or ruled out).
- The PCR AMGX/AMGY design has been shown to not only[clarification needed] facilitate in amplifying DNA sequences from a very minuscule amount of genome. However it can also be used for real-time sex determination from forensic bone samples. This provides a powerful and effective way to determine gender in forensic cases and ancient specimens.[42]
Research applications[edit]
PCR has been applied to many areas of research in molecular genetics:
- PCR allows rapid production of short pieces of DNA, even when not more than the sequence of the two primers is known. This ability of PCR augments many methods, such as generating hybridization probes for Southern or northern blot hybridization. PCR supplies these techniques with large amounts of pure DNA, sometimes as a single strand, enabling analysis even from very small amounts of starting material.
- The task of DNA sequencing can also be assisted by PCR. Known segments of DNA can easily be produced from a patient with a genetic disease mutation. Modifications to the amplification technique can extract segments from a completely unknown genome, or can generate just a single strand of an area of interest.
- PCR has numerous applications to the more traditional process of DNA cloning. It can extract segments for insertion into a vector from a larger genome, which may be only available in small quantities. Using a single set of ‘vector primers’, it can also analyze or extract fragments that have already been inserted into vectors. Some alterations to the PCR protocol can generate mutations (general or site-directed) of an inserted fragment.
- Sequence-tagged sites is a process where PCR is used as an indicator that a particular segment of a genome is present in a particular clone. The Human Genome Project found this application vital to mapping the cosmid clones they were sequencing, and to coordinating the results from different laboratories.
- An application of PCR is the phylogenic analysis of DNA from ancient sources, such as that found in the recovered bones of Neanderthals, from frozen tissues of mammoths, or from the brain of Egyptian mummies.[16] In some cases the highly degraded DNA from these sources might be reassembled during the early stages of amplification.
- A common application of PCR is the study of patterns of gene expression. Tissues (or even individual cells) can be analyzed at different stages to see which genes have become active, or which have been switched off. This application can also use quantitative PCR to quantitate the actual levels of expression
- The ability of PCR to simultaneously amplify several loci from individual sperm[43] has greatly enhanced the more traditional task of genetic mapping by studying chromosomal crossovers after meiosis. Rare crossover events between very close loci have been directly observed by analyzing thousands of individual sperms. Similarly, unusual deletions, insertions, translocations, or inversions can be analyzed, all without having to wait (or pay) for the long and laborious processes of fertilization, embryogenesis, etc.
- Site-directed mutagenesis: PCR can be used to create mutant genes with mutations chosen by scientists at will. These mutations can be chosen in order to understand how proteins accomplish their functions, and to change or improve protein function.
Advantages[edit]
PCR has a number of advantages. It is fairly simple to understand and to use, and produces results rapidly. The technique is highly sensitive with the potential to produce millions to billions of copies of a specific product for sequencing, cloning, and analysis. qRT-PCR shares the same advantages as the PCR, with an added advantage of quantification of the synthesized product. Therefore, it has its uses to analyze alterations of gene expression levels in tumors, microbes, or other disease states.[27]
PCR is a very powerful and practical research tool. The sequencing of unknown etiologies of many diseases are being figured out by the PCR. The technique can help identify the sequence of previously unknown viruses related to those already known and thus give us a better understanding of the disease itself. If the procedure can be further simplified and sensitive non-radiometric detection systems can be developed, the PCR will assume a prominent place in the clinical laboratory for years to come.[16]
Limitations[edit]
One major limitation of PCR is that prior information about the target sequence is necessary in order to generate the primers that will allow its selective amplification.[27] This means that, typically, PCR users must know the precise sequence(s) upstream of the target region on each of the two single-stranded templates in order to ensure that the DNA polymerase properly binds to the primer-template hybrids and subsequently generates the entire target region during DNA synthesis.
Like all enzymes, DNA polymerases are also prone to error, which in turn causes mutations in the PCR fragments that are generated.[44]
Another limitation of PCR is that even the smallest amount of contaminating DNA can be amplified, resulting in misleading or ambiguous results. To minimize the chance of contamination, investigators should reserve separate rooms for reagent preparation, the PCR, and analysis of product. Reagents should be dispensed into single-use aliquots. Pipettors with disposable plungers and extra-long pipette tips should be routinely used.[16] It is moreover recommended to ensure that the lab set-up follows a unidirectional workflow. No materials or reagents used in the PCR and analysis rooms should ever be taken into the PCR preparation room without thorough decontamination.[45]
Environmental samples that contain humic acids may inhibit PCR amplification and lead to inaccurate results.
Variations[edit]
- Allele-specific PCR or The amplification refractory mutation system (ARMS): a diagnostic or cloning technique based on single-nucleotide variations (SNVs not to be confused with SNPs) (single-base differences in a patient). Any mutation involving single base change can be detected by this system. It requires prior knowledge of a DNA sequence, including differences between alleles, and uses primers whose 3′ ends encompass the SNV (base pair buffer around SNV usually incorporated).[46] PCR amplification under stringent conditions is much less efficient in the presence of a mismatch between template and primer, so successful amplification with an SNP-specific primer signals presence of the specific SNP or small deletions in a sequence.[47] See SNP genotyping for more information.
- Assembly PCR or Polymerase Cycling Assembly (PCA): artificial synthesis of long DNA sequences by performing PCR on a pool of long oligonucleotides with short overlapping segments. The oligonucleotides alternate between sense and antisense directions, and the overlapping segments determine the order of the PCR fragments, thereby selectively producing the final long DNA product.[48]
- Asymmetric PCR: preferentially amplifies one DNA strand in a double-stranded DNA template. It is used in sequencing and hybridization probing where amplification of only one of the two complementary strands is required. PCR is carried out as usual, but with a great excess of the primer for the strand targeted for amplification. Because of the slow (arithmetic) amplification later in the reaction after the limiting primer has been used up, extra cycles of PCR are required.[49] A recent modification on this process, known as Linear-After-The-Exponential-PCR (LATE-PCR), uses a limiting primer with a higher melting temperature (Tm) than the excess primer to maintain reaction efficiency as the limiting primer concentration decreases mid-reaction.[50]
- Convective PCR: a pseudo-isothermal way of performing PCR. Instead of repeatedly heating and cooling the PCR mixture, the solution is subjected to a thermal gradient. The resulting thermal instability driven convective flow automatically shuffles the PCR reagents from the hot and cold regions repeatedly enabling PCR.[51] Parameters such as thermal boundary conditions and geometry of the PCR enclosure can be optimized to yield robust and rapid PCR by harnessing the emergence of chaotic flow fields.[52] Such convective flow PCR setup significantly reduces device power requirement and operation time.
- Dial-out PCR: a highly parallel method for retrieving accurate DNA molecules for gene synthesis. A complex library of DNA molecules is modified with unique flanking tags before massively parallel sequencing. Tag-directed primers then enable the retrieval of molecules with desired sequences by PCR.[53]
- Digital PCR (dPCR): used to measure the quantity of a target DNA sequence in a DNA sample. The DNA sample is highly diluted so that after running many PCRs in parallel, some of them do not receive a single molecule of the target DNA. The target DNA concentration is calculated using the proportion of negative outcomes. Hence the name ‘digital PCR’.
- Helicase-dependent amplification: similar to traditional PCR, but uses a constant temperature rather than cycling through denaturation and annealing/extension cycles. DNA helicase, an enzyme that unwinds DNA, is used in place of thermal denaturation.[54]
- Hot start PCR: a technique that reduces non-specific amplification during the initial set up stages of the PCR. It may be performed manually by heating the reaction components to the denaturation temperature (e.g., 95 °C) before adding the polymerase.[55] Specialized enzyme systems have been developed that inhibit the polymerase’s activity at ambient temperature, either by the binding of an antibody[13][56] or by the presence of covalently bound inhibitors that dissociate only after a high-temperature activation step. Hot-start/cold-finish PCR is achieved with new hybrid polymerases that are inactive at ambient temperature and are instantly activated at elongation temperature.
- In silico PCR (digital PCR, virtual PCR, electronic PCR, e-PCR) refers to computational tools used to calculate theoretical polymerase chain reaction results using a given set of primers (probes) to amplify DNA sequences from a sequenced genome or transcriptome. In silico PCR was proposed as an educational tool for molecular biology.[57]
- Intersequence-specific PCR (ISSR): a PCR method for DNA fingerprinting that amplifies regions between simple sequence repeats to produce a unique fingerprint of amplified fragment lengths.[58]
- Inverse PCR: is commonly used to identify the flanking sequences around genomic inserts. It involves a series of DNA digestions and self ligation, resulting in known sequences at either end of the unknown sequence.[59]
- Ligation-mediated PCR: uses small DNA linkers ligated to the DNA of interest and multiple primers annealing to the DNA linkers; it has been used for DNA sequencing, genome walking, and DNA footprinting.[60]
- Methylation-specific PCR (MSP): developed by Stephen Baylin and James G. Herman at the Johns Hopkins School of Medicine,[61] and is used to detect methylation of CpG islands in genomic DNA. DNA is first treated with sodium bisulfite, which converts unmethylated cytosine bases to uracil, which is recognized by PCR primers as thymine. Two PCRs are then carried out on the modified DNA, using primer sets identical except at any CpG islands within the primer sequences. At these points, one primer set recognizes DNA with cytosines to amplify methylated DNA, and one set recognizes DNA with uracil or thymine to amplify unmethylated DNA. MSP using qPCR can also be performed to obtain quantitative rather than qualitative information about methylation.
- Miniprimer PCR: uses a thermostable polymerase (S-Tbr) that can extend from short primers («smalligos») as short as 9 or 10 nucleotides. This method permits PCR targeting to smaller primer binding regions, and is used to amplify conserved DNA sequences, such as the 16S (or eukaryotic 18S) rRNA gene.[62]
- Multiplex ligation-dependent probe amplification (MLPA): permits amplifying multiple targets with a single primer pair, thus avoiding the resolution limitations of multiplex PCR (see below).
- Multiplex-PCR: consists of multiple primer sets within a single PCR mixture to produce amplicons of varying sizes that are specific to different DNA sequences. By targeting multiple genes at once, additional information may be gained from a single test-run that otherwise would require several times the reagents and more time to perform. Annealing temperatures for each of the primer sets must be optimized to work correctly within a single reaction, and amplicon sizes. That is, their base pair length should be different enough to form distinct bands when visualized by gel electrophoresis.
- Nanoparticle-assisted PCR (nanoPCR): some nanoparticles (NPs) can enhance the efficiency of PCR (thus being called nanoPCR), and some can even outperform the original PCR enhancers. It was reported that quantum dots (QDs) can improve PCR specificity and efficiency. Single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs) are efficient in enhancing the amplification of long PCR. Carbon nanopowder (CNP) can improve the efficiency of repeated PCR and long PCR, while zinc oxide, titanium dioxide and Ag NPs were found to increase the PCR yield. Previous data indicated that non-metallic NPs retained acceptable amplification fidelity. Given that many NPs are capable of enhancing PCR efficiency, it is clear that there is likely to be great potential for nanoPCR technology improvements and product development.[63][64]
- Nested PCR: increases the specificity of DNA amplification, by reducing background due to non-specific amplification of DNA. Two sets of primers are used in two successive PCRs. In the first reaction, one pair of primers is used to generate DNA products, which besides the intended target, may still consist of non-specifically amplified DNA fragments. The product(s) are then used in a second PCR with a set of primers whose binding sites are completely or partially different from and located 3′ of each of the primers used in the first reaction. Nested PCR is often more successful in specifically amplifying long DNA fragments than conventional PCR, but it requires more detailed knowledge of the target sequences.
- Overlap-extension PCR or Splicing by overlap extension (SOEing) : a genetic engineering technique that is used to splice together two or more DNA fragments that contain complementary sequences. It is used to join DNA pieces containing genes, regulatory sequences, or mutations; the technique enables creation of specific and long DNA constructs. It can also introduce deletions, insertions or point mutations into a DNA sequence.[65][66]
- PAN-AC: uses isothermal conditions for amplification, and may be used in living cells.[67][68]
- Quantitative PCR (qPCR): used to measure the quantity of a target sequence (commonly in real-time). It quantitatively measures starting amounts of DNA, cDNA, or RNA. Quantitative PCR is commonly used to determine whether a DNA sequence is present in a sample and the number of its copies in the sample. Quantitative PCR has a very high degree of precision. Quantitative PCR methods use fluorescent dyes, such as Sybr Green, EvaGreen or fluorophore-containing DNA probes, such as TaqMan, to measure the amount of amplified product in real time. It is also sometimes abbreviated to RT-PCR (real-time PCR) but this abbreviation should be used only for reverse transcription PCR. qPCR is the appropriate contractions for quantitative PCR (real-time PCR).
- Reverse Complement PCR (RC-PCR): Allows the addition of functional domains or sequences of choice to be appended independently to either end of the generated amplicon in a single closed tube reaction. This method generates target specific primers within the reaction by the interaction of universal primers (which contain the desired sequences or domains to be appended) and RC probes.
- Reverse Transcription PCR (RT-PCR): for amplifying DNA from RNA. Reverse transcriptase reverse transcribes RNA into cDNA, which is then amplified by PCR. RT-PCR is widely used in expression profiling, to determine the expression of a gene or to identify the sequence of an RNA transcript, including transcription start and termination sites. If the genomic DNA sequence of a gene is known, RT-PCR can be used to map the location of exons and introns in the gene. The 5′ end of a gene (corresponding to the transcription start site) is typically identified by RACE-PCR (Rapid Amplification of cDNA Ends).
- RNase H-dependent PCR (rhPCR): a modification of PCR that utilizes primers with a 3′ extension block that can be removed by a thermostable RNase HII enzyme. This system reduces primer-dimers and allows for multiplexed reactions to be performed with higher numbers of primers.[69]
- Single specific primer-PCR (SSP-PCR): allows the amplification of double-stranded DNA even when the sequence information is available at one end only. This method permits amplification of genes for which only a partial sequence information is available, and allows unidirectional genome walking from known into unknown regions of the chromosome.[70]
- Solid Phase PCR: encompasses multiple meanings, including Polony Amplification (where PCR colonies are derived in a gel matrix, for example), Bridge PCR[71] (primers are covalently linked to a solid-support surface), conventional Solid Phase PCR (where Asymmetric PCR is applied in the presence of solid support bearing primer with sequence matching one of the aqueous primers) and Enhanced Solid Phase PCR[72] (where conventional Solid Phase PCR can be improved by employing high Tm and nested solid support primer with optional application of a thermal ‘step’ to favour solid support priming).
- Suicide PCR: typically used in paleogenetics or other studies where avoiding false positives and ensuring the specificity of the amplified fragment is the highest priority. It was originally described in a study to verify the presence of the microbe Yersinia pestis in dental samples obtained from 14th Century graves of people supposedly killed by the plague during the medieval Black Death epidemic.[73] The method prescribes the use of any primer combination only once in a PCR (hence the term «suicide»), which should never have been used in any positive control PCR reaction, and the primers should always target a genomic region never amplified before in the lab using this or any other set of primers. This ensures that no contaminating DNA from previous PCR reactions is present in the lab, which could otherwise generate false positives.
- Thermal asymmetric interlaced PCR (TAIL-PCR): for isolation of an unknown sequence flanking a known sequence. Within the known sequence, TAIL-PCR uses a nested pair of primers with differing annealing temperatures; a degenerate primer is used to amplify in the other direction from the unknown sequence.[74]
- Touchdown PCR (Step-down PCR): a variant of PCR that aims to reduce nonspecific background by gradually lowering the annealing temperature as PCR cycling progresses. The annealing temperature at the initial cycles is usually a few degrees (3–5 °C) above the Tm of the primers used, while at the later cycles, it is a few degrees (3–5 °C) below the primer Tm. The higher temperatures give greater specificity for primer binding, and the lower temperatures permit more efficient amplification from the specific products formed during the initial cycles.[75]
- Universal Fast Walking: for genome walking and genetic fingerprinting using a more specific ‘two-sided’ PCR than conventional ‘one-sided’ approaches (using only one gene-specific primer and one general primer—which can lead to artefactual ‘noise’)[76] by virtue of a mechanism involving lariat structure formation. Streamlined derivatives of UFW are LaNe RAGE (lariat-dependent nested PCR for rapid amplification of genomic DNA ends),[77] 5’RACE LaNe[78] and 3’RACE LaNe.[79]
History[edit]
Diagrammatic representation of an example primer pair. The use of primers in an in vitro assay to allow DNA synthesis was a major innovation that allowed the development of PCR.
The heat-resistant enzymes that are a key component in polymerase chain reaction were discovered in the 1960s as a product of a microbial life form that lived in the superheated waters of Yellowstone’s Mushroom Spring.[80]
A 1971 paper in the Journal of Molecular Biology by Kjell Kleppe and co-workers in the laboratory of H. Gobind Khorana first described a method of using an enzymatic assay to replicate a short DNA template with primers in vitro.[81] However, this early manifestation of the basic PCR principle did not receive much attention at the time and the invention of the polymerase chain reaction in 1983 is generally credited to Kary Mullis.[82][page needed]
«Baby Blue», a 1986 prototype machine for doing PCR
When Mullis developed the PCR in 1983, he was working in Emeryville, California for Cetus Corporation, one of the first biotechnology companies, where he was responsible for synthesizing short chains of DNA. Mullis has written that he conceived the idea for PCR while cruising along the Pacific Coast Highway one night in his car.[83] He was playing in his mind with a new way of analyzing changes (mutations) in DNA when he realized that he had instead invented a method of amplifying any DNA region through repeated cycles of duplication driven by DNA polymerase. In Scientific American, Mullis summarized the procedure: «Beginning with a single molecule of the genetic material DNA, the PCR can generate 100 billion similar molecules in an afternoon. The reaction is easy to execute. It requires no more than a test tube, a few simple reagents, and a source of heat.»[84] DNA fingerprinting was first used for paternity testing in 1988.[85]
Mullis has credited his use of LSD as integral to his development of PCR: «Would I have invented PCR if I hadn’t taken LSD? I seriously doubt it. I could sit on a DNA molecule and watch the polymers go by. I learnt that partly on psychedelic drugs.»[86]
Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA,[1] were jointly awarded the Nobel Prize in Chemistry in 1993, seven years after Mullis and his colleagues at Cetus first put his proposal to practice.[87] Mullis’s 1985 paper with R. K. Saiki and H. A. Erlich, «Enzymatic Amplification of β-globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia»—the polymerase chain reaction invention (PCR)—was honored by a Citation for Chemical Breakthrough Award from the Division of History of Chemistry of the American Chemical Society in 2017.[88][2]
At the core of the PCR method is the use of a suitable DNA polymerase able to withstand the high temperatures of >90 °C (194 °F) required for separation of the two DNA strands in the DNA double helix after each replication cycle. The DNA polymerases initially employed for in vitro experiments presaging PCR were unable to withstand these high temperatures.[2] So the early procedures for DNA replication were very inefficient and time-consuming, and required large amounts of DNA polymerase and continuous handling throughout the process.
The discovery in 1976 of Taq polymerase—a DNA polymerase purified from the thermophilic bacterium, Thermus aquaticus, which naturally lives in hot (50 to 80 °C (122 to 176 °F)) environments[14] such as hot springs—paved the way for dramatic improvements of the PCR method. The DNA polymerase isolated from T. aquaticus is stable at high temperatures remaining active even after DNA denaturation,[15] thus obviating the need to add new DNA polymerase after each cycle.[3] This allowed an automated thermocycler-based process for DNA amplification.
Patent disputes[edit]
The PCR technique was patented by Kary Mullis and assigned to Cetus Corporation, where Mullis worked when he invented the technique in 1983. The Taq polymerase enzyme was also covered by patents. There have been several high-profile lawsuits related to the technique, including an unsuccessful lawsuit brought by DuPont. The Swiss pharmaceutical company Hoffmann-La Roche purchased the rights to the patents in 1992. The last of the commercial PCR patents expired in 2017.[89]
A related patent battle over the Taq polymerase enzyme is still ongoing[as of?] in several jurisdictions around the world between Roche and Promega. The legal arguments have extended beyond the lives of the original PCR and Taq polymerase patents, which expired on 28 March 2005.[90]
See also[edit]
- COVID-19 testing
- DNA spiking
- Loop-mediated isothermal amplification
- Selector technique
- Thermus thermophilus
- Pfu DNA polymerase
References[edit]
- ^ a b «The Nobel Prize in Chemistry 1993». NobelPrize.org.
- ^ a b c d Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N (December 1985). «Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia». Science. 230 (4732): 1350–54. Bibcode:1985Sci…230.1350S. doi:10.1126/science.2999980. PMID 2999980.
- ^ a b Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, et al. (January 1988). «Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase». Science. 239 (4839): 487–91. Bibcode:1988Sci…239..487S. doi:10.1126/science.239.4839.487. PMID 2448875.
- ^ Enners, Edward; Porta, Angela R. (2012). «Determining Annealing Temperatures for Polymerase Chain Reaction». The American Biology Teacher. 74 (4): 256–60. doi:10.1525/abt.2012.74.4.9. S2CID 86708426.
- ^ a b c d e f Ninfa, Alexander; Ballou, David; Benore, Marilee (2009). Fundamental Laboratory Approaches for Biochemistry and Biotechnology. United States: Wiley. pp. 408–10. ISBN 978-0-470-08766-4.
- ^ Cheng S, Fockler C, Barnes WM, Higuchi R (June 1994). «Effective amplification of long targets from cloned inserts and human genomic DNA». Proceedings of the National Academy of Sciences of the United States of America. 91 (12): 5695–99. Bibcode:1994PNAS…91.5695C. doi:10.1073/pnas.91.12.5695. PMC 44063. PMID 8202550.
- ^ Carr AC, Moore SD (2012). Lucia A (ed.). «Robust quantification of polymerase chain reactions using global fitting». PLOS ONE. 7 (5): e37640. Bibcode:2012PLoSO…737640C. doi:10.1371/journal.pone.0037640. PMC 3365123. PMID 22701526.
- ^ a b Joseph Sambrook & David W. Russel (2001). Molecular Cloning: A Laboratory Manual (third ed.). Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. ISBN 978-0-879-69576-7. Chapter 8: In vitro Amplification of DNA by the Polymerase Chain Reaction
- ^ «Polymerase Chain Reaction (PCR)». National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ «PCR». Genetic Science Learning Center, University of Utah.
- ^ Pavlov AR, Pavlova NV, Kozyavkin SA, Slesarev AI (May 2004). «Recent developments in the optimization of thermostable DNA polymerases for efficient applications». Trends in Biotechnology. 22 (5): 253–60. doi:10.1016/j.tibtech.2004.02.011. PMID 15109812.
- ^ Rychlik W, Spencer WJ, Rhoads RE (November 1990). «Optimization of the annealing temperature for DNA amplification in vitro». Nucleic Acids Research. 18 (21): 6409–12. doi:10.1093/nar/18.21.6409. PMC 332522. PMID 2243783.
- ^ a b Sharkey DJ, Scalice ER, Christy KG, Atwood SM, Daiss JL (May 1994). «Antibodies as thermolabile switches: high temperature triggering for the polymerase chain reaction». Bio/Technology. 12 (5): 506–09. doi:10.1038/nbt0594-506. PMID 7764710. S2CID 2885453.
- ^ a b Chien A, Edgar DB, Trela JM (September 1976). «Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus». Journal of Bacteriology. 127 (3): 1550–57. doi:10.1128/jb.127.3.1550-1557.1976. PMC 232952. PMID 8432.
- ^ a b Lawyer FC, Stoffel S, Saiki RK, Chang SY, Landre PA, Abramson RD, Gelfand DH (May 1993). «High-level expression, purification, and enzymatic characterization of full-length Thermus aquaticus DNA polymerase and a truncated form deficient in 5′ to 3′ exonuclease activity». PCR Methods and Applications. 2 (4): 275–87. doi:10.1101/gr.2.4.275. PMID 8324500.
- ^ a b c d Schochetman G, Ou CY, Jones WK (December 1988). «Polymerase chain reaction». The Journal of Infectious Diseases. 158 (6): 1154–57. doi:10.1093/infdis/158.6.1154. JSTOR 30137034. PMID 2461996.
- ^ Borman, Jon; Schuster, David; Li, Wu-bo; Jessee, Joel; Rashtchian, Ayoub (2000). «PCR from problematic templates» (PDF). Focus. 22 (1): 10. Archived from the original (PDF) on 7 March 2017.
- ^ Bogetto, Prachi; Waidne, Lisa; Anderson, Holly (2000). «Helpful tips for PCR» (PDF). Focus. 22 (1): 12. Archived from the original (PDF) on 7 March 2017.
- ^ Biolabs, New England. «Q5® High-Fidelity DNA Polymerase | NEB». www.neb.com. Retrieved 4 December 2021.
- ^ Sze, Marc A.; Schloss, Patrick D. (2019). «The Impact of DNA Polymerase and Number of Rounds of Amplification in PCR on 16S rRNA Gene Sequence Data». mSphere. 4 (3): e00163–19. doi:10.1128/mSphere.00163-19. PMC 6531881. PMID 31118299.
- ^ Sarkar G, Kapelner S, Sommer SS (December 1990). «Formamide can dramatically improve the specificity of PCR». Nucleic Acids Research. 18 (24): 7465. doi:10.1093/nar/18.24.7465. PMC 332902. PMID 2259646.
- ^ «Electronic PCR». NCBI – National Center for Biotechnology Information. Retrieved 13 March 2012.
- ^ Pavlov AR, Pavlova NV, Kozyavkin SA, Slesarev AI (2006). «Thermostable DNA Polymerases for a Wide Spectrum of Applications: Comparison of a Robust Hybrid TopoTaq to other enzymes». In Kieleczawa J (ed.). DNA Sequencing II: Optimizing Preparation and Cleanup. Jones & Bartlett. pp. 241–57. ISBN 978-0-7637-3383-4.
- ^ Pombert JF, Sistek V, Boissinot M, Frenette M (October 2009). «Evolutionary relationships among salivarius streptococci as inferred from multilocus phylogenies based on 16S rRNA-encoding, recA, secA, and secY gene sequences». BMC Microbiology. 9: 232. doi:10.1186/1471-2180-9-232. PMC 2777182. PMID 19878555.
- ^ «Chemical Synthesis, Sequencing, and Amplification of DNA (class notes on MBB/BIO 343)». Arizona State University. Archived from the original on 9 October 1997. Retrieved 29 October 2007.
- ^ Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. (April 2009). «The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments» (PDF). Clinical Chemistry. 55 (4): 611–22. doi:10.1373/clinchem.2008.112797. PMID 19246619.
- ^ a b c d Garibyan L, Avashia N (March 2013). «Polymerase chain reaction». The Journal of Investigative Dermatology. 133 (3): 1–4. doi:10.1038/jid.2013.1. PMC 4102308. PMID 23399825.
- ^ Schnell, S.; Mendoza, C. (October 1997). «Theoretical Description of the Polymerase Chain Reaction». Journal of Theoretical Biology. 188 (3): 313–18. Bibcode:1997JThBi.188..313S. doi:10.1006/jtbi.1997.0473. PMID 9344735.
- ^ Schnell, S.; Mendoza, C. (21 February 1997). «Enzymological Considerations for the Theoretical Description of the Quantitative Competitive Polymerase Chain Reaction (QC-PCR)». Journal of Theoretical Biology. 184 (4): 433–40. Bibcode:1997JThBi.184..433S. doi:10.1006/jtbi.1996.0283. ISSN 0022-5193. PMID 9082073.
- ^ Becker, Sven; Böger, Peter; Oehlmann, Ralfh; Ernst, Anneliese (1 November 2000). «PCR Bias in Ecological Analysis: a Case Study for Quantitative Taq Nuclease Assays in Analyses of Microbial Communities». Applied and Environmental Microbiology. 66 (11): 4945–53. Bibcode:2000ApEnM..66.4945B. doi:10.1128/AEM.66.11.4945-4953.2000. ISSN 1098-5336. PMC 92404. PMID 11055948.
- ^ Solomon, Anthony W.; Peeling, Rosanna W.; Foster, Allen; Mabey, David C. W. (1 October 2004). «Diagnosis and Assessment of Trachoma». Clinical Microbiology Reviews. 17 (4): 982–1011. doi:10.1128/CMR.17.4.982-1011.2004. ISSN 0893-8512. PMC 523557. PMID 15489358.
- ^ Ramzy, Reda M.R. (April 2002). «Recent advances in molecular diagnostic techniques for human lymphatic filariasis and their use in epidemiological research». Transactions of the Royal Society of Tropical Medicine and Hygiene. 96: S225–29. doi:10.1016/S0035-9203(02)90080-5. PMID 12055843.
- ^ Sachse, Konrad (2003). «Specificity and performance of diagnostic PCR assays». In Sachse, Konrad; Frey, Joachim (eds.). PCR Detection of Microbial Pathogens. Methods in Molecular Biology. Vol. 216. Totowa, New Jersey: Humana Press. pp. 3–29. doi:10.1385/1-59259-344-5:03. ISBN 978-1-59259-344-6. PMID 12512353.
- ^ Quill E (March 2008). «Medicine. Blood-matching goes genetic». Science. 319 (5869): 1478–79. doi:10.1126/science.319.5869.1478. PMID 18339916. S2CID 36945291.
- ^ Tomar, Rukam (2010). Molecular Markers and Plant Biotechnology. Pitman Pura, New Delhi: New India Publishing Agency. p. 188. ISBN 978-93-80235-25-7.
- ^ a b c Cai HY, Caswell JL, Prescott JF (March 2014). «Nonculture molecular techniques for diagnosis of bacterial disease in animals: a diagnostic laboratory perspective». Veterinary Pathology. 51 (2): 341–50. doi:10.1177/0300985813511132. PMID 24569613.
- ^ Salis AD (2009). «Applications in Clinical Microbiology». Real-Time PCR: Current Technology and Applications. Caister Academic Press. ISBN 978-1-904455-39-4.
- ^ Kwok S, Mack DH, Mullis KB, Poiesz B, Ehrlich G, Blair D, et al. (May 1987). «Identification of human immunodeficiency virus sequences by using in vitro enzymatic amplification and oligomer cleavage detection». Journal of Virology. 61 (5): 1690–94. doi:10.1128/jvi.61.5.1690-1694.1987. PMC 254157. PMID 2437321.
- ^ «Coronavirus: il viaggio dei test». Istituto Superiore di Sanità.
- ^ Finger, Horst; von Koenig, Carl Heinz Wirsing (1996). Baron, Samuel (ed.). Medical Microbiology (4th ed.). Galveston, TX: University of Texas Medical Branch at Galveston. ISBN 978-0-9631172-1-2. PMID 21413270.
- ^ Yeh, Sylvia H.; Mink, ChrisAnna M. (2012). «Bordetella pertussis and Pertussis (Whooping Cough)». Netter’s Infectious Diseases. Netter’s Infectious Diseases. pp. 11–14. doi:10.1016/B978-1-4377-0126-5.00003-3. ISBN 978-1-4377-0126-5.
- ^ Alonso A, Martín P, Albarrán C, García P, García O, de Simón LF, et al. (January 2004). «Real-Time PCR Designs to Estimate Nuclear and Mitochondrial DNA Copy Number in Forensic and Ancient DNA Studies». Forensic Science International. 139 (2–3): 141–49. doi:10.1016/j.forsciint.2003.10.008. PMID 15040907.
- ^ Boehnke M, Arnheim N, Li H, Collins FS (July 1989). «Fine-structure genetic mapping of human chromosomes using the polymerase chain reaction on single sperm: experimental design considerations». American Journal of Human Genetics. 45 (1): 21–32. PMC 1683385. PMID 2568090.
- ^ Zhou YH, Zhang XP, Ebright RH (November 1991). «Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase». Nucleic Acids Research. 19 (21): 6052. doi:10.1093/nar/19.21.6052. PMC 329070. PMID 1658751.
- ^ Stursberg, Stephanie (2021). «Setting up a PCR lab from scratch». INTEGRA Biosciences.
{{cite web}}: CS1 maint: url-status (link) - ^ Bulduk, BK, et al. (March 2020). «A Novel Amplification-Refractory Mutation System-PCR Strategy to Screen MT-TL1 Pathogenic Variants in Patient Repositories». Genet Test Mol Biomarkers. 24 (3): 165–170. doi:10.1089/gtmb.2019.0079. PMID 32167396.
- ^ Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, et al. (April 1989). «Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS)». Nucleic Acids Research. 17 (7): 2503–16. doi:10.1093/nar/17.7.2503. PMC 317639. PMID 2785681.
- ^ Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL (October 1995). «Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides». Gene. 164 (1): 49–53. doi:10.1016/0378-1119(95)00511-4. PMID 7590320.
- ^ Innis MA, Myambo KB, Gelfand DH, Brow MA (December 1988). «DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA». Proceedings of the National Academy of Sciences of the United States of America. 85 (24): 9436–40. Bibcode:1988PNAS…85.9436I. doi:10.1073/pnas.85.24.9436. PMC 282767. PMID 3200828.
- ^ Pierce KE, Wangh LJ (2007). Linear-after-the-exponential polymerase chain reaction and allied technologies Real-time detection strategies for rapid, reliable diagnosis from single cells. Methods in Molecular Medicine. Vol. 132. pp. 65–85. doi:10.1007/978-1-59745-298-4_7. ISBN 978-1-58829-578-1. PMID 17876077.
- ^ Krishnan M, Ugaz VM, Burns MA (October 2002). «PCR in a Rayleigh-Bénard convection cell». Science. 298 (5594): 793. doi:10.1126/science.298.5594.793. PMID 12399582.
- ^ Priye A, Hassan YA, Ugaz VM (November 2013). «Microscale chaotic advection enables robust convective DNA replication». Analytical Chemistry. 85 (21): 10536–41. doi:10.1021/ac402611s. PMID 24083802.
- ^ Schwartz JJ, Lee C, Shendure J (September 2012). «Accurate gene synthesis with tag-directed retrieval of sequence-verified DNA molecules». Nature Methods. 9 (9): 913–15. doi:10.1038/nmeth.2137. PMC 3433648. PMID 22886093.
- ^ Vincent M, Xu Y, Kong H (August 2004). «Helicase-dependent isothermal DNA amplification». EMBO Reports. 5 (8): 795–800. doi:10.1038/sj.embor.7400200. PMC 1249482. PMID 15247927.
- ^ Chou Q, Russell M, Birch DE, Raymond J, Bloch W (April 1992). «Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications». Nucleic Acids Research. 20 (7): 1717–23. doi:10.1093/nar/20.7.1717. PMC 312262. PMID 1579465.
- ^ Kellogg DE, Rybalkin I, Chen S, Mukhamedova N, Vlasik T, Siebert PD, Chenchik A (June 1994). «TaqStart Antibody: «hot start» PCR facilitated by a neutralizing monoclonal antibody directed against Taq DNA polymerase». BioTechniques. 16 (6): 1134–37. PMID 8074881.
- ^ San Millán RM, Martínez-Ballesteros I, Rementeria A, Garaizar J, Bikandi J (December 2013). «Online exercise for the design and simulation of PCR and PCR-RFLP experiments». BMC Research Notes. 6: 513. doi:10.1186/1756-0500-6-513. PMC 4029544. PMID 24314313.
- ^ Zietkiewicz E, Rafalski A, Labuda D (March 1994). «Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification». Genomics. 20 (2): 176–83. doi:10.1006/geno.1994.1151. PMID 8020964.
- ^ Ochman H, Gerber AS, Hartl DL (November 1988). «Genetic applications of an inverse polymerase chain reaction». Genetics. 120 (3): 621–23. doi:10.1093/genetics/120.3.621. PMC 1203539. PMID 2852134.
- ^ Mueller PR, Wold B (November 1989). «In vivo footprinting of a muscle specific enhancer by ligation mediated PCR». Science. 246 (4931): 780–86. Bibcode:1989Sci…246..780M. doi:10.1126/science.2814500. PMID 2814500.
- ^ Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (September 1996). «Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands». Proceedings of the National Academy of Sciences of the United States of America. 93 (18): 9821–26. Bibcode:1996PNAS…93.9821H. doi:10.1073/pnas.93.18.9821. PMC 38513. PMID 8790415.
- ^ Isenbarger TA, Finney M, Ríos-Velázquez C, Handelsman J, Ruvkun G (February 2008). «Miniprimer PCR, a new lens for viewing the microbial world». Applied and Environmental Microbiology. 74 (3): 840–49. Bibcode:2008ApEnM..74..840I. doi:10.1128/AEM.01933-07. PMC 2227730. PMID 18083877.
- ^ Shen C, Yang W, Ji Q, Maki H, Dong A, Zhang Z (November 2009). «NanoPCR observation: different levels of DNA replication fidelity in nanoparticle-enhanced polymerase chain reactions». Nanotechnology. 20 (45): 455103. Bibcode:2009Nanot..20S5103S. doi:10.1088/0957-4484/20/45/455103. PMID 19822925. S2CID 3393115.
- ^ Shen, Cenchao (2013). «An Overview of Nanoparticle-Assisted Polymerase Chain Reaction Technology». An Overview of Nanoparticle‐Assisted Polymerase Chain Reaction Technology. US: Wiley-Blackwell Publishing Ltd. pp. 97–106. doi:10.1002/9781118451915.ch5. ISBN 978-1-118-45191-5.
- ^ Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (April 1989). «Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension». Gene. 77 (1): 61–68. doi:10.1016/0378-1119(89)90359-4. PMID 2744488.
- ^ Moller, Simon (2006). PCR: The Basics. US: Taylor & Francis Group. p. 144. ISBN 978-0-415-35547-6.
- ^ David F, Turlotte E (November 1998). «[A method of isothermal gene amplification]» [An Isothermal Amplification Method]. Comptes Rendus de l’Académie des Sciences, Série III. 321 (11): 909–14. Bibcode:1998CRASG.321..909D. doi:10.1016/S0764-4469(99)80005-5. PMID 9879470.
- ^ Fabrice David (September–October 2002). «Utiliser les propriétés topologiques de l’ADN: une nouvelle arme contre les agents pathogènes» (PDF). Fusion. Archived from the original (PDF) on 28 November 2007.(in French)
- ^ Dobosy JR, Rose SD, Beltz KR, Rupp SM, Powers KM, Behlke MA, Walder JA (August 2011). «RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers». BMC Biotechnology. 11: 80. doi:10.1186/1472-6750-11-80. PMC 3224242. PMID 21831278.
- ^ Shyamala, V.; Ferro-Luzzi, Ames G. (1993). Single Specific Primer-Polymerase Chain Reaction (SSP-PCR) and Genome Walking. Methods in Molecular Biology. Vol. 15. pp. 339–48. doi:10.1385/0-89603-244-2:339. ISBN 978-0-89603-244-6. PMID 21400290.
- ^ Bing DH, Boles C, Rehman FN, Audeh M, Belmarsh M, Kelley B, Adams CP (1996). «Bridge amplification: a solid phase PCR system for the amplification and detection of allelic differences in single copy genes». Genetic Identity Conference Proceedings, Seventh International Symposium on Human Identification. Archived from the original on 7 May 2001.
- ^ Khan Z, Poetter K, Park DJ (April 2008). «Enhanced solid phase PCR: mechanisms to increase priming by solid support primers». Analytical Biochemistry. 375 (2): 391–93. doi:10.1016/j.ab.2008.01.021. PMID 18267099.
- ^ Raoult D, Aboudharam G, Crubézy E, Larrouy G, Ludes B, Drancourt M (November 2000). «Molecular identification by «suicide PCR» of Yersinia pestis as the agent of medieval black death». Proceedings of the National Academy of Sciences of the United States of America. 97 (23): 12800–03. Bibcode:2000PNAS…9712800R. doi:10.1073/pnas.220225197. PMC 18844. PMID 11058154.
- ^ Liu YG, Whittier RF (February 1995). «Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking». Genomics. 25 (3): 674–81. doi:10.1016/0888-7543(95)80010-J. PMID 7759102.
- ^ Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (July 1991). «‘Touchdown’ PCR to circumvent spurious priming during gene amplification». Nucleic Acids Research. 19 (14): 4008. doi:10.1093/nar/19.14.4008. PMC 328507. PMID 1861999.
- ^ Myrick KV, Gelbart WM (February 2002). «Universal Fast Walking for direct and versatile determination of flanking sequence». Gene. 284 (1–2): 125–31. doi:10.1016/S0378-1119(02)00384-0. PMID 11891053.
- ^ «Full Text – LaNe RAGE: a new tool for genomic DNA flanking sequence determination». www.ejbiotechnology.info.
- ^ Park DJ (January 2005). «A new 5′ terminal murine GAPDH exon identified using 5’RACE LaNe». Molecular Biotechnology. 29 (1): 39–46. doi:10.1385/MB:29:1:39. PMID 15668518. S2CID 45702164.
- ^ Park DJ (April 2004). «3′ RACE LaNe: a simple and rapid fully nested PCR method to determine 3′-terminal cDNA sequence». BioTechniques. 36 (4): 586–88, 590. doi:10.2144/04364BM04. PMID 15088375.
- ^ «Key ingredient in coronavirus tests comes from Yellowstone’s lakes». Science. 31 March 2020. Retrieved 13 May 2020.
- ^ Kleppe K, Ohtsuka E, Kleppe R, Molineux I, Khorana HG (March 1971). «Studies on polynucleotides. XCVI. Repair replications of short synthetic DNA’s as catalyzed by DNA polymerases». Journal of Molecular Biology. 56 (2): 341–61. doi:10.1016/0022-2836(71)90469-4. PMID 4927950.
- ^ Rabinow, Paul (1996). Making PCR: A Story of Biotechnology. Chicago: University of Chicago Press. ISBN 978-0-226-70146-2.
- ^ Mullis, Kary (1998). Dancing Naked in the Mind Field. New York: Pantheon Books. ISBN 978-0-679-44255-4.
- ^ Mullis KB (April 1990). «The unusual origin of the polymerase chain reaction». Scientific American. 262 (4): 56–61, 64–65. Bibcode:1990SciAm.262d..56M. doi:10.1038/scientificamerican0490-56. PMID 2315679.
- ^ Patidar M, Agrawal S, Parveen F, Khare P (2015). «Molecular insights of saliva in solving paternity dispute». Journal of Forensic Dental Sciences. 7 (1): 76–79. doi:10.4103/0975-1475.150325. PMC 4330625. PMID 25709326.
- ^ Nichols D, Barker E (2016). «Psychedelics». Pharmacological Reviews. 68 (2): 264–355. doi:10.1124/pr.115.011478. PMC 4813425. PMID 26841800.
- ^ «The Nobel Prize in Chemistry 1993». NobelPrize.org.
- ^ «Citations for Chemical Breakthrough Awards 2017 Awardees». Division of the History of Chemistry. Retrieved 12 March 2018.
- ^ Fore, J. Jr.; Wiechers, I. R.; Cook-Deegan, R. (3 July 2006). «The effects of business practices, licensing, and intellectual property on development and dissemination of the polymerase chain reaction: case study». Journal of Biomedical Discovery and Collaboration. 1: 7. doi:10.1186/1747-5333-1-7. PMC 1523369. PMID 16817955.
- ^ «Advice on How to Survive the Taq Wars». GEN Genetic Engineering News – Biobusiness Channel. 26 (9). 1 May 2006.
External links[edit]
- US Patent for PCR
- What is PCR plateau effect? YouTube tutorial video
- History of the Polymerase Chain Reaction from the Smithsonian Institution Archives
Опубликовано: 10.02.2021 13:07:00 Обновлено: 18.02.2021 Просмотров: 257885

ПЦР – высокоточный метод диагностики и одно из самых главных открытий в области биологии за последние десятилетия. ПЦР-анализ применяется уже почти 40 лет и считается наиболее точным и чувствительным способом диагностики инфекционных заболеваний.
ПЦР – уникальный и универсальный метод, тест используется не только в клинической лабораторной диагностике, но также в биологии, криминалистике, археологии и многих других научных областях. Все, что нужно для проведения анализа – небольшое количество любого биоматериала пациента.
Суть метода ПЦР
ПЦР – полимеразная цепная реакция. Метод основан на обнаружении даже небольших концентраций искомого элемента диагностики. Для определения изначально крайне малых концентраций РНК или ДНК, которые необходимо определить в процессе проведения основного этапа исследования, используется метод искусственного увеличения количества РНК или ДНК. А поскольку они специфичны и строго индивидуальны для каждого микроорганизма или живого существа за счет уникальности последовательности нуклеотидов во фрагментах, ошибка в определении целевого ДНК или РНК исключена.
Генетическая информация любого живого организма записывается в ДНК. Эта молекула состоит из двух цепочек, сплетающихся в единую спираль. Некоторые вирусы (например, COVID-19) хранят свой код в РНК – одной нити нуклеотидов.
Для каждого организма, включая вирусы, бактерии и грибки, последовательность нуклеотидов уникальна. Ее можно сравнить с отпечатком пальца или сканом сетчатки глаза человека. Укороченные последовательности нуклеотидов, характерные для каждого вида патогена (возбудителя опасных заболеваний), хранятся в базах научных лабораторий в виде праймеров – отдельных участков ДНК, типичных для только конкретного возбудителя. Эти участки значительно короче любой молекулы ДНК. Такие праймеры присоединяются к ДНК возбудителя в пробе и под действием катализаторов многократно воспроизводят свои дубли. Этот процесс называются «репликация» – многократное увеличение, дублирование искомого участка до тех пор, пока он не станет доступен для определения. Процесс репликации возможен только при наличии в пробе ДНК возбудителя.
Преимущества метода ПЦР
- Высокая специфичность. Метод ПЦР определяет заданную последовательность нуклеотидов, присущую конкретному патогену. Таким образом, специфичность теста стремится к 100%. Исключен риск ложноположительных результатов.
- Чувствительность. Для ПЦР достаточно всего несколько молекул ДНК патогена (или даже уже неактивных – разрушенных вирусных частиц, сохранивших специфические участки ДНК в достаточном количестве), чтобы он был обнаружен в ходе исследования.
- Скорость проведения. Лаборатория получает результат ПЦР-теста через несколько часов, клиент – уже на следующий день. Скорость диагностики имеет принципиально важное значение для своевременного лечения. Например, при диагностике бактериальных инфекций классический посев занимает от нескольких дней, а с ПЦР-анализом пациент сможет принять меры и начать лечение уже через сутки.
- Универсальность. Для исследования подходит любой биоматериал: кровь, моча, сперма, мокрота, гной, жидкости из абсцессов и пр. Кроме этого, ПЦР применяется в самых разных областях науки и медицины, работая даже там, где другие методы бессильны.
- Диагностика латентных инфекций. ПЦР-диагностика определяет возбудителя инфекции даже в инкубационном периоде и при скрытом течении заболевания.
Какие есть недостатки
Единственный серьезный недостаток, связанный с методом полимеразной цепной реакции, — его высокая технологичность. Исследования ПЦР требуют строжайших соблюдений правил и серьезной оснащенности лабораторного комплекса. Не каждая лаборатория может позволить себе все необходимое оборудование.
Показания к проведению ПЦР-анализа
ПЦР – один из наиболее востребованных способов диагностики в медицине, и применяется он в самых разных областях:
- Диагностика инфекционных заболеваний (гепатит, ВИЧ, TORCH-инфекции и огромное множество других видов патогенов).
- Урогинекология (диагностика инфекций, передающихся половым путем: хламидиоз, уреаплазма, микоплазма, кандиды, гарднерелла, герпесвирусы). Важное значение для женского и мужского здоровья играет ВПЧ – вирус папилломы человека. Доказано, что у женщин онкогенные штаммы этого вируса способны вызывать рак шейки матки.
- Неонатология. Существует целый ряд инфекций, способных поражать плод еще в материнской утробе. Среди них вирусы герпеса, токсоплазмы, краснухи. ПЦР-диагностика позволяет правильно определить тактику ведения и риски внутриутробной инфекции.
- Респираторные заболевания. Диагностика методом ПЦР стала в 2020 году актуальной как никогда и продемонстрировала свою незаменимость и эффективность. ПЦР-анализ – главный тест и «золотой стандарт» диагностики коронавирусной инфекции Sars-Cov-2 (COVID-19), ставшей причиной самой масштабной пандемии последних десятилетий.
- Генетика. Наследственные заболевания и отцовство также диагностируются при помощи метода ПЦР.
ПЦР-тесты применяются везде, где нужен быстрый, точный и надежный результат.
Подготовка к проведению ПЦР-теста
Еще одно немаловажное преимущество ПЦР-анализов – это отсутствие специфической подготовки к проведению тестов. Достаточно следовать стандартным рекомендациям специалиста:
- Анализ сдается утром натощак. При этом ряд генетических исследований проводится в произвольное время, удобное пациенту.
- При сдаче анализа по мазку из ротоглотки необходимо выдержать интервал с приемом пищи и воды в 3-4 часа перед тестом.
- Перед анализом на венерические инфекции необходимо воздержаться от половой активности в течение суток.
- Перед анализом нельзя использовать никакие противовирусные препараты.
Подробную инструкцию по правилам подготовки к каждому анализу вы всегда можете получить у лечащего врача или у консультантов на нашей горячей линии.
Как проводится ПЦР-анализ
С 1983 года, когда был изобретен данный метод, прошло много времени, и технологии не стоят на месте. Сегодня существует несколько различных методик для проведения ПЦР-теста:
- С обратной транскрипцией. Самый распространенный способ идентификации известной последовательности РНК, включающий амплификацию, определение патогена и его идентификации среди образцов, хранящихся в научной картотеке.
- Вложенная ПЦР (или «гнездовая») — используется для снижения количества неспецифичных продуктов реакции и имеет две стадии с использованием двух видов праймеров.
- Изотермические методы – не требуют повторяющихся температурных циклов, менее энергозатратны.
- Инвертированная ПЦР — применяется, если имеется только короткий фрагмент известной последовательности, но необходимо определить соседние последовательности после вставки ДНК в геном.
- ПЦР в реальном времени — метод, позволяющий определить не только присутствие целевой нуклеотидной последовательности в образце, но и измерять количество ее копий после каждого цикла амплификации, что дает возможность для проведения тестов с количественным результатом
Это далеко не все существующие на сегодняшний день разновидности ПЦР-технологий. Ученые активно развивают эту важную и перспективную область лабораторной диагностики, совершенствуют методы ПЦР, оттачивают техники и изобретают новые подходы.
Результаты ПЦР-теста
Результаты анализов, проведенных методом ПЦР, известны уже через один день. Иногда возможно проведение экстренного теста – его часто используют при оказании срочной медицинской помощи при госпитализации. Тогда срок готовности результата сокращается до считанных часов.
Результаты ПЦР-теста дадут точную информацию о том, какая инфекция была обнаружена. При количественном тестировании анализ определит также вирусную или бактериальную нагрузку на организм. В этом случае в результатах будет значиться титр обнаруженного патогена (его количество в одном миллилитре пробы). Количественный анализ особенно важен при диагностике заболеваний, спровоцированных условно-патогенными микроорганизмами, которые присутствуют в норме практически у каждого человека. Такие микроорганизмы представляют угрозу только при большой численности, а в остальных случаях мирно сосуществуют с носителем.
Метод ПЦР

Полимеразная цепная реакция (ПЦР) — высокоточный метод диагностики заболеваний, основанный на копировании ДНК или РНК патогена в пробе.
СОДЕРЖАНИЕ
Какие инфекции позволяет выявить ПЦР
Как подготовиться к сдаче анализа методом ПЦР
Что такое ПЦР
Полимеразная цепная реакция — высокоточный метод молекулярной биологии, который позволяет обнаружить в биоматериале ДНК и РНК патогенов. Отличается чувствительностью и специфичностью, а также скоростью получения результата.
В последнее время метод часто используется для диагностики новой коронавирусной инфекции COVID-19.
Полимеразную цепную реакцию изобрёл учёный-биохимик Кэри Муллис в 1983 году. Через 10 лет он получил за это Нобелевскую премию по химии.
Как работает ПЦР
В ходе исследования пробирку с биоматериалом помещают в особые условия, под воздействием которых молекулы нуклеиновой кислоты (двухнитевая ДНК, присутствующая в пробе или синтезированная из однонитевой РНК), кодирующие генетическую информацию определённого патогена, многократно копируются.
Если в исследуемом образце было хотя бы небольшое количество патогенных частиц, то через несколько часов их число увеличится до десятков и даже сотен миллионов. Это позволяет легко обнаружить возбудителя инфекции или генетическую «поломку» при очень низких рисках ложноотрицательного результата.

Молекула ДНК
Разновидности ПЦР
ПЦР — перспективное направление исследований. Учёные постоянно совершенствуют способы проведения реакции и изобретают новые методики, адаптируя ПЦР под разные цели.
В лабораторной диагностике применяются два основных метода — ПЦР в режиме реального времени и ПЦР с обратной транскрипцией.
С помощью ПЦР в режиме реального времени (количественной) можно не только выявить патогенные ДНК-молекулы, но и подсчитать количество их копий. Это позволяет рассчитать степень инфицирования, дозировку лекарств и длительность их приёма.
ПЦР с обратной транскрипцией применяется, если в качестве искомого генетического материала выступает не ДНК, а РНК вируса.
Дело в том, что у некоторых патогенов, например у вируса гепатита C, генетический материал представлен только РНК — однонитевой цепочкой нуклеотидов, в отличие от двухцепочечной ДНК-молекулы. Она тоже несёт в себе генетическую информацию, но не подходит для стандартной ПЦР.
Чтобы провести исследование, РНК-молекуле «восстанавливают» вторую нить с помощью особого фермента — обратной транскриптазы. Так РНК превращается в ДНК-молекулу, готовую к дальнейшему копированию с помощью стандартной ПЦР или ПЦР в режиме реального времени.
Как происходит ПЦР
Что нужно для ПЦР
Для проведения полимеразной цепной реакции используется специальный прибор — амплификатор. Он поддерживает необходимую для каждого этапа температуру и проводит циклы копирования ДНК — амплификацию.
Когда молекул становится достаточно для распознавания, оборудование анализирует их, а затем выдает качественный или количественный результат.
Пробирки с биоматериалом, подготовленным для ПЦР, в амплификаторе
Помимо амплификатора, для ПЦР необходимы определённые компоненты:
- Целевая ДНК — частица ДНК искомого патогена. Она состоит из уникальной последовательности нуклеотидов. Например, при ПЦР-анализе на герпес целью служит та часть ДНК, которая отличает вирус герпеса от других вирусов. Целевую ДНК также называют ДНК-матрицей.
Нити ДНК и РНК состоят из маленьких звеньев — нуклеотидов. Их последовательность образует генетический код, несущий наследственную информацию.
- Два праймера — короткие нити из нуклеотидов, которые подходят к искомым нитям ДНК, как детали пазла. На основе праймеров в ходе ПЦР создаются новые ДНК-молекулы. Если праймеры не подошли, значит, целевой ДНК в биоматериале нет.
- ДНК-полимераза — фермент, который достраивает нить ДНК в ходе ПЦР.
- Дезоксинуклеотидтрифосфаты (дНТФ) — смесь нуклеотидов, строительный материал для копий ДНК-молекул.
- Ионы магния (Mg2+) — поддерживают активность ДНК-полимеразы.
- Буферный раствор — смесь, которая создаёт оптимальные условия для биохимической реакции.
Этапы полимеразной цепной реакции
ПЦР состоит из трёх основных этапов.
Сначала пробу нагревают, и все ДНК-молекулы, которые есть в биоматериале, распадаются на две нити РНК. Этот процесс называется денатурацией.
Если генетический материал патогена — РНК, а не ДНК, то перед денатурацией проводят ПЦР с обратной транскрипцией. Этот метод позволяет превратить РНК в ДНК-молекулу.
Затем температуру снижают. Если в пробе есть целевая ДНК, праймеры присоединяются к концам двух её нитей — так происходит отжиг.
После отжига фермент ДНК-полимераза активируется и начинает достраивать вторую нить ДНК, начиная от праймера. Для восстановления цепи ДНК полимераза использует дНТФ. Заключительный этап называют элонгацией.
При проведении ПЦР выполняют более 30 циклов копирования (амплификации). В результате каждого цикла количество ДНК-цепей удваивается. Так, если на первом цикле было 2 цепи ДНК, на втором их будет уже 4, а на третьем — 8, и так далее.
Какие инфекции позволяет выявить ПЦР
Каждый патоген — бактерия, вирус или гриб — содержит уникальные РНК- или ДНК-молекулы.
По наличию этих молекул в биоматериале можно быстро и точно диагностировать заражение болезнетворными микроорганизмами. Но есть важное условие: у ПЦР-исследования должна быть цель — конкретный патоген, наличие которого нужно подтвердить или опровергнуть. Как правило, цель ПЦР определяет врач на основании осмотра, инструментального обследования и жалоб пациента.
Вирусные инфекции
У любых вирусов есть ДНК- или РНК-молекулы. ПЦР позволяет обнаружить их в организме, а ПЦР в реальном времени — ещё и подсчитать количество амплифицированных копий. Результат ПЦР может помочь врачу поставить диагноз, определить стадию болезни, подобрать дозировку лекарств и определить длительность курса лечения.
28.93. Соскоб (+450 ₽)  561 1 день
561 1 день
Соскоб (+450 ₽) 1 день
 561 бонус на счёт
561 бонус на счёт
28.95. Соскоб (+450 ₽) 646 2 дня
Соскоб (+450 ₽) 2 дня
646 бонусов на счёт
28.548. Вен. кровь (+220 ₽) 425 3 дня
Вен. кровь (+220 ₽) 3 дня
425 бонусов на счёт
19.67.2. Соскоб (+450 ₽) 52 Колич. 1 день
Соскоб (+450 ₽) Колич. 1 день
52 бонуса на счёт
Бактериальные инфекции
Как и у вирусов, у всех бактерий есть ДНК-молекулы, которые можно выявить с помощью ПЦР.
19.21.1. Соскоб (+450 ₽) 46 Кач. 1 день
Соскоб (+450 ₽) Кач. 1 день
46 бонусов на счёт
19.50.1. Соскоб (+450 ₽) 49 Кач. 2 дня
Соскоб (+450 ₽) Кач. 2 дня
49 бонусов на счёт
19.20.1. Соскоб (+450 ₽) 46 Кач. 1 день
Соскоб (+450 ₽) Кач. 1 день
46 бонусов на счёт
19.56.1. Вен. кровь (+220 ₽) 53 2 дня
Вен. кровь (+220 ₽) 2 дня
53 бонуса на счёт
28.95. Соскоб (+450 ₽) 646 2 дня
Соскоб (+450 ₽) 2 дня
646 бонусов на счёт
Но не все бактерии, в отличие от вирусов, вредны. Например, условно-патогенные бактерии в небольших количествах приносят пользу организму. Однако, если по каким-либо причинам их становится слишком много, возникает бактериальный дисбаланс — дисбактериоз. Это состояние вредит здоровью.
Чаще всего условно-патогенные бактерии становятся причиной кожных, кишечных и гинекологических заболеваний. Для их диагностики используют качественную ПЦР и ПЦР в режиме реального времени — она позволяет выявить, превысило ли количество бактерий норму, и если да, то насколько сильно.
Опираясь на результат такого анализа, врач может рассчитать дозировку и длительность применения лекарств, оптимальную для восстановления нормальной микрофлоры.
19.60.1. Соскоб (+450 ₽) 63 1 день
Соскоб (+450 ₽) 1 день
63 бонуса на счёт
19.61.1. Соскоб (+450 ₽) 47 Колич. 1 день
Соскоб (+450 ₽) Колич. 1 день
47 бонусов на счёт
19.63.1. Соскоб (+450 ₽) 47 Колич. 1 день
Соскоб (+450 ₽) Колич. 1 день
47 бонусов на счёт
27.39. Соскоб (+450 ₽) 358 3 дня
Соскоб (+450 ₽) 3 дня
358 бонусов на счёт

28.118. Соскоб (+450 ₽) 300 3 дня
Соскоб (+450 ₽) 3 дня
300 бонусов на счёт
28.98.1. Соскоб (+450 ₽) 175 1 день
Соскоб (+450 ₽) 1 день
175 бонусов на счёт
Грибковые инфекции
В большинстве случаев для диагностики грибковых инфекций используется микроскопия: визуально обнаружить грибок не составляет труда, особенно при наличии выраженных симптомов.
Однако в некоторых случаях, например при кандидозе (молочнице), широко применяется ПЦР. Это связано с двумя основными факторами.
Во-первых, возбудитель кандидоза(кандида) — условно-патогенный грибок. Она становится болезнетворной, только если её слишком много.
ПЦР в режиме реального времени позволяет уточнить, насколько сильно «разрослась» кандида, и подобрать оптимальную дозировку препарата, чтобы сократить её количество.
Во-вторых, слизистые оболочки урогенитального тракта, влагалища и рта густо населены различными микроорганизмами. ПЦР в таком случае — более точный метод диагностики благодаря своей прицельности.
19.22.1. Соскоб (+450 ₽) 46 Кач. 1 день
Соскоб (+450 ₽) Кач. 1 день
46 бонусов на счёт
19.66.1. Соскоб (+450 ₽) 47 Колич. 1 день
Соскоб (+450 ₽) Колич. 1 день
47 бонусов на счёт
19.22.2. 46 Колич. 1 день
Колич. 1 день
46 бонусов на счёт
19.66.2. 47 1 день
1 день
47 бонусов на счёт
Где ещё применяют ПЦР
Поскольку генетический код есть у всех живых существ, ПЦР-метод применяется везде, где может понадобиться многократное увеличение числа ДНК- и РНК-молекул. Например, для получения пригодного для дальнейших исследований генетического материала с места преступления или поиска конкретных генетических мутаций.
ПЦР в установлении родства
Если биоматериала недостаточно для определения родства, перед основным анализом может потребоваться амплификация молекул ДНК. Большое количество биоматериала позволит провести более точное исследование. Например, установить пол, вид или родство как живых, так и мёртвых существ.
ПЦР в диагностике предрасположенностей
С помощью ПЦР можно выявить определённые гены, которые отвечают за предрасположенность человека к различным состояниям (например, к избыточному весу) или болезням.
Подобные исследования назначаются, если пациенту нужен план профилактики развития заболеваний.
GNP184 Взятие (2 вида, +580 ₽) 980 5 дней
Взятие (2 вида, +580 ₽) 5 дней
980 бонусов на счёт

GNP082 Взятие (2 вида, +580 ₽) 274 3 дня
Взятие (2 вида, +580 ₽) 3 дня
274 бонуса на счёт
ПЦР в диагностике генетических мутаций
Ещё полимеразная цепная реакция позволяет обнаруживать в генах мутации, которые могут влиять на эффективность лечения различных заболеваний. Например, в случае бронхиальной астмы или сердечной недостаточности результат исследования на ген бета-2-адренорецептора ADRB2 может помочь врачу подобрать лекарства.
GN0011 Вен. кровь (+220 ₽) 109 Кач. 3 дня
Вен. кровь (+220 ₽) Кач. 3 дня
109 бонусов на счёт
Кроме того, некоторые мутации в генах могут служить маркерами онкологических заболеваний. Их обнаружение позволяет специалистам подобрать наиболее эффективную терапию и спрогнозировать дальнейшее развитие патологий.
GNP095 1 321 10 дней
10 дней
1 321 бонус на счёт
GNP100 Вен. кровь (+220 ₽) 870 Кач. 12 дней
Вен. кровь (+220 ₽) Кач. 12 дней
870 бонусов на счёт
GNP085 Выделение ДНК (+360 ₽) 352 14 дней
Выделение ДНК (+360 ₽) 14 дней
352 бонуса на счёт
GNP106 Вен. кровь (+220 ₽) 1 013 32 дня
Вен. кровь (+220 ₽) 32 дня
1 013 бонусов на счёт
GNP077 800 Кач. 7 дней
Кач. 7 дней
800 бонусов на счёт
GN0071 Взятие (2 вида, +580 ₽) 109 Кач. 3 дня
Взятие (2 вида, +580 ₽) Кач. 3 дня
109 бонусов на счёт
ПЦР в судебно-медицинской экспертизе и криминалистике
Более узкая сфера для применения генетических ПЦР-тестов — расследования.
Почти 99,9% ДНК разных людей идентичны. Однако оставшаяся 0,1% — это уникальная часть генетического кода, которая отличает людей друг от друга. Выделение и сравнение этих уникальных частей позволяет идентифицировать преступника или жертву.
ПЦР может применяться в тех случаях, когда генетического материала слишком мало для точного исследования. Например, если были найдены разложившиеся останки тела или оно сильно обгорело, и опознать его невозможно.
В случае поиска преступника генетический материал может помочь доказать вину подозреваемого. ПЦР позволяет выделить ДНК из любых биологических «следов», которые злоумышленник мог оставить. Подойдут кровь, слюна, моча, волосы, семенная жидкость, частицы кожи под ногтями жертвы. Выделить ДНК можно даже из отпечатка пальца.
Отпечаток пальца содержит около 10 нанограмм ДНК. Для исследования достаточно 0,1 нанограмма.
ПЦР в ветеринарии
Ветеринары используют ПЦР так же, как и врачи: для диагностики у животных различных инфекционных заболеваний, а также оценки эффективности лечения.
У диких и бродячих животных ПЦР помогает выявить патогены, которые передаются людям и вызывают опасные для жизни заболевания. Например, бешенство, бубонную чуму, некоторые виды гриппа, оспу и другие.
19.53. 122 Кач. 2 дня
Кач. 2 дня
122 бонуса на счёт
50.8.2090. Соскоб (+450 ₽) 326 2 дня
Соскоб (+450 ₽) 2 дня
326 бонусов на счёт
19.107. Вен. кровь (+220 ₽) 144 2 дня
Вен. кровь (+220 ₽) 2 дня
144 бонуса на счёт
19.39.1. Вен. кровь (+220 ₽) 48 1 день
Вен. кровь (+220 ₽) 1 день
48 бонусов на счёт
ПЦР в растениеводстве
Для выявления фитопатологий — заболеваний растений — полимеразная цепная реакция тоже применяется. Она позволяет обнаружить вирусы, бактерии и грибы, которые могут привести к частичной или полной гибели урожая.
Проблемой применения ПЦР в растениеводстве служит только стоимость оборудования, которое не все хозяйства могут себе позволить, а также высокая чувствительность метода. Дело в том, что если образец загрязнён незначительным количеством патогена, результат анализа будет положительным.
Эта особенность требует более тщательной подготовки растительного материала к исследованию, а также стерильных условий в лаборатории и дополнительных проверочных тестов.
ПЦР в контроле потребительских продуктов
ПЦР может использоваться для контроля применения генно-модифицированных организмов (ГМО), которые часто присутствуют в продуктах питания и различных кормах.
Генно-модифицированные организмы создаются с помощью генной инженерии — «редактирования» исходного генетического кода организма и «прививания» ему полезных свойств. Это позволяет приспособить растения к неблагоприятному климату, вредителям или химикатам, а также заставить их быстрее расти или приносить больше плодов.
ПЦР позволяет многократно копировать удачную ДНК-молекулу, выявлять ГМО, а также создавать миллионы образцов для дальнейшего применения или для новых экспериментов и разработок.
Преимущества ПЦР
Полимеразная цепная реакция считается одним из самых эффективных способов выявить инфицирование. Метод точен — он позволяет обнаружить искомый патоген, даже если его концентрация в пробе крайне мала.
Кроме того, результат ПЦР не зависит от иммунной реакции организма, культивационных особенностей патогена или стадии инфицирования.
Точность
Точность ПЦР складывается из двух критериев — чувствительности и специфичности.
Чем ниже вероятность ложноотрицательного результата, тем выше чувствительность метода.
Чувствительность ПЦР составляет 90–100% — с такой вероятностью анализ выявит патоген, если он действительно есть в пробе.
По сравнению с полимеразной цепной реакцией, другие методы менее чувствительны. Так, например, чувствительность иммуноферментного анализа составляет 50–70%, а культурального исследования — 60–80%.
Чем ниже вероятность ложноположительного результата, тем выше специфичность метода.
Специфичность ПЦР достигает 100%. Если в биоматериале нет подходящей РНК или ДНК, реакция не произойдёт.
Высокой специфичностью обладает и культуральный метод, однако он менее чувствителен, и это снижает его общую точность.
Иммуноферментный анализ, в отличие от ПЦР и культурального метода, выявляет не патоген, а антитела — особые белки, которые иммунная система вырабатывает для борьбы с ним.
После болезни антитела могут давать перекрёстную реакцию с другими антителами. Например, при анализе на болезнь Лайма положительный результат могут дать антитела к возбудителю инфекционного мононуклеоза и другим герпес-вирусам. В обоих случаях результат может быть ложным, так как фактически пациент не заражён искомым патогеном и/или заражён чем-то другим.
Таким образом, высокая чувствительность и специфичность полимеразной цепной реакции делают её одним из самых точных методов лабораторной диагностики.
Скорость
В большинстве случаев результат ПЦР можно получить через один день.
Срок готовности посева (культуральный метод) — в среднем от двух-трёх дней.
Готовность результата иммуноферментного анализа — в среднем около двух дней.
Независимость от особенностей патогена
По сравнению с другими методами лабораторной диагностики, на успех ПЦР не влияют некоторые характеристики патогенов.
Так, например, суть культурального метода (посева) заключается в выращивании бактерий и грибов на комфортной для них питательной среде.
Если в пробе были микроорганизмы, со временем их численность увеличится и станет заметна даже без микроскопа.
Однако некоторые бактерии культивировать сложно. Например, боррелии и бледная трепонема настолько требовательны к условиям питательной среды и посева, что этот метод невозможно применять в лабораторной диагностике болезни Лайма и сифилиса.
Риккетсии и хламидии — внутриклеточные паразиты, они в целом не выращиваются в лабораторных условиях, а шигеллы и сальмонеллы не всегда активны и в определённых фазах не размножаются.
27.39. Соскоб (+450 ₽) 358 3 дня
Соскоб (+450 ₽) 3 дня
358 бонусов на счёт
19.50.1. Соскоб (+450 ₽) 49 Кач. 2 дня
Соскоб (+450 ₽) Кач. 2 дня
49 бонусов на счёт
19.56.1. Вен. кровь (+220 ₽) 53 2 дня
Вен. кровь (+220 ₽) 2 дня
53 бонуса на счёт
На эффективность ПЦР особенности выращивания грибов и бактерий не влияют. Более того, с помощью ПЦР можно проводить исследование ещё и на вирусы.
Колонии бактерий, выращенные культуральным методом
Независимость от иммунной реакции и наличия симптомов
Серологические исследования опираются на наличие у больного антител к патогенам. Если заражение произошло недавно, инфекция протекает в скрытой форме или у пациента есть иммунные нарушения, результат серологического исследования может быть ложным.
Так, например, первые антитела к возбудителю гепатита B появляются только через 1–3 месяца. Соответственно, ранняя диагностика болезни с помощью анализа на антитела будет неэффективна.
Кроме того, иммунная система некоторых пациентов вообще не вырабатывает антитела при подтверждённой ПЦР инфекции. Такое бывает, например, при клещевом энцефалите.
ПЦР, напротив, не зависит от иммунной реакции, и с её помощью можно диагностировать заболевание в самом начале — до появления антител и первых симптомов.
Около 50% серологических исследований на гепатит B в инкубационном и начальном периоде дают ложноотрицательный результат при положительной ПЦР.
Как и при многих других инфекционных заболеваниях, при гепатите ранняя диагностика играет важную роль. Она позволяет сразу начать лечение, минимизировать последствия болезни и предотвратить дальнейшее распространение возбудителя. В таких случаях ПЦР — предпочтительный метод исследования.
19.44. Вен. кровь (+220 ₽) 440 3 дня
Вен. кровь (+220 ₽) 3 дня
440 бонусов на счёт
19.3. Вен. кровь (+220 ₽) 92 Кач. 3 дня
Вен. кровь (+220 ₽) Кач. 3 дня
92 бонуса на счёт
Недостатки ПЦР
Точечное применение
ПЦР должна иметь одну определённую цель — патоген, генетический материал которого будет копироваться.
Если у пациента микст-инфекция (заражение сразу несколькими патогенами), ПЦР-исследование обнаружит только тот патоген, на который ориентирован анализ. Обнаружить дополнительные вирусы или бактерии заодно с целевым инфекционным агентом не получится.
Чувствительность оборудования
Оборудование для ПЦР очень чувствительное и требует стерильности как в процедурном кабинете, так и в самой лаборатории.
До 70% ошибочных результатов ПЦР связаны с нарушением правил подготовки проб.
Если проба случайно загрязнится в процессе взятия, транспортировки или добавления реагентов, результат исследования может быть ложным.
Как подготовиться к сдаче анализа методом ПЦР
Основная задача при взятии биоматериала для ПЦР — не загрязнить его. Для этого нужно строго соблюдать правила подготовки.
Подготовка к ПЦР-анализу крови
- Кровь следует сдавать натощак, с 8 до 11 часов утра. В течение дня показатели крови могут существенно меняться, результат утреннего анализа — самый достоверный.
- За 24 часа до теста следует исключить алкоголь и воздержаться от интенсивных физических нагрузок.
- За 8 часов до взятия крови не следует есть, а также пить соки, молоко или другие напитки. Можно пить негазированную воду. Накануне исследования лучше поужинать лёгкой, нежирной пищей.
- За 1–2 часа до анализа желательно не курить, избегать стресса и физического напряжения (бег, быстрый подъём по лестнице).
- За 15 минут до взятия крови желательно немного отдохнуть: посидеть в лабораторном отделении, отдышаться, успокоиться.
- За 1–2 дня до исследования исключить из рациона продукты с высоким содержанием жиров. За 2 дня до сдачи крови на вирусные гепатиты исключить из рациона цитрусовые, оранжевые фрукты и овощи.
Правила взятия крови у детей:
- младенцы до 1 года
- дети до 7 лет
Для контроля показателей в динамике следует сдавать анализ в одинаковых условиях: в той же лаборатории, в то же время суток.
Лекарства и медицинские процедуры могут повлиять на результат анализа.
Не следует сдавать кровь сразу после физиотерапевтических процедур, инструментального обследования, рентгенологического или ультразвукового исследования, массажа.
Лучше всего выполнять исследование крови до начала приёма лекарственных препаратов или через 10–14 дней после их отмены. О принимаемых лекарствах следует предупредить медсестру, а также врача, который выполняет диагностику или назначает лечение.
Исследования для оценки эффективности лечения, как правило, проводят через 1–2 недели после последнего приёма препарата.
Подготовка к ПЦР-анализу мочи
За 24 часа до анализа следует исключить из рациона продукты, меняющие цвет и химический состав мочи: алкогольные напитки, «красящие» овощи — свёклу и морковь, а также специи и соль. По согласованию с врачом лучше отказаться от приёма мочегонных препаратов.
Перед взятием биоматериала необходимо тщательно вымыть наружные половые органы.
Женщинам рекомендуется обработать половые губы ватными тампонами: сначала — смоченными в тёплой воде с мылом, затем — смоченными в чистой воде.
Движения тампона должны быть направлены от лобка к заднему проходу. Лишнюю влагу удалить чистой салфеткой.
Мужчинам следует тщательно вымыть отверстие мочеиспускательного канала тёплой водой с мылом, оттянув кожную складку, промыть водой и промокнуть чистой салфеткой.
Биоматериал следует собрать в одноразовый стерильный контейнер. Его можно бесплатно получить в любом лабораторном отделении Гемотест.
Инструкция по использованию контейнера
- Открутите крышку контейнера и положите её, не касаясь внутренней части крышки.
- Соберите первую порцию мочи в контейнер.
- Если мочу на анализ берут у ребёнка, можно использовать мочеприёмник.
- Закройте контейнер крышкой и плотно закрутите её.
- Доставьте контейнер в лабораторное отделение в течение 2 часов после взятия биоматериала.
Подготовка к ПЦР-анализу кала
Кал следует сдавать до приёма антибиотиков или не ранее, чем через 12 часов после приёма препарата.
За 72 часа до исследования откажитесь от использования ректальных свечей и масел, слабительных средств и клизм, а также прекратите приём препаратов, оказывающих влияние на работу кишечника и окраску кала.
Соберите образец кала после естественного опорожнения кишечника без применения клизм или слабительных средств.
Позаботьтесь о том, чтобы в биоматериал не попали посторонние примеси: вода, моча, выделения половых органов, дезинфицирующие средства и химические вещества, содержащиеся в подгузниках:
- перед дефекацией вымойте и высушите промежность;
- соберите кал с чистой и не впитывающей влагу поверхности: это может быть чистый пакет из полиэтилена, клеёнка, чистое судно или горшок.
Собирать кал из унитаза, пелёнок или подгузников — нельзя!
Поместите 1–2 чайные ложки биоматериала в герметичный контейнер. Если нужно выполнить несколько анализов, подготовьте для каждого исследования отдельную порцию биоматериала в отдельном контейнере.
Доставьте контейнер в лабораторное отделение в течение 2 часов после сбора. Чтобы сохранить температуру контейнера, можно поместить его в термос с кубиком льда или в пакет с хладоэлементами.
Если отслеживаете показатели в динамике, сдавайте каждый анализ при одинаковых условиях: в том же лабораторном отделении, тем же методом.
Исследования для оценки эффективности лечения, как правило, проводят через 2–4 недели после приёма антибактериальных препаратов.
Подготовка к ПЦР-анализу слюны
За 24 часа до исследования по предварительному согласованию с лечащим врачом следует отказаться от терапии полости рта какими-либо лекарственными препаратами.
За 3 часа до исследования рекомендуется отказаться от курения, употребления пищи и гигиены полости рта (полоскание, чистка зубов, применение освежителей для рта, употребление леденцов, жевательной резинки).
Подготовка к ПЦР-анализу мокроты
- За 2 недели до исследования по согласованию с лечащим врачом следует прекратить приём противомикробных препаратов и иммуномодуляторов.
- Мокроту следует собирать утром, сразу после пробуждения. Предварительно нужно почистить зубы и прополоскать рот, нельзя есть до взятия биоматериала.
- Важно собирать не слюну или носоглоточную слизь, а мокроту. Сделайте несколько глубоких вдохов, чтобы вызвать «мокрый» кашель. Необходимо откашлять содержимое глубоких дыхательных путей.
- Сплюньте мокроту в заранее подготовленный стерильный контейнер с плотно закрывающейся крышкой. Минимальный объём мокроты — 1 мл.
- Доставьте контейнер в лабораторное отделение в день взятия биоматериала.
Подготовка к соскобу из зева
За 24 часа до исследования по предварительному согласованию с лечащим врачом следует отказаться от терапии полости рта какими-либо лекарственными препаратами.
За 3 часа до исследования рекомендуется отказаться от курения, употребления пищи и гигиены полости рта (полоскание, чистка зубов, применение освежителей для рта, употребление леденцов, жевательной резинки).
Подготовка к соскобу из влагалища или урогенитального тракта
За 30 дней до исследования следует прекратить применение антибиотиков по согласованию с лечащим врачом.
За 24 часа до исследования не следует проводить спринцевание, внутривагинальную терапию (свечи, тампоны), не использовать антисептические средства (в том числе мирамистин), исключить половой контакт.
В день исследования запрещается проводить туалет половых органов.
Женщинам рекомендуется сдавать анализ до менструации или через 5 дней после её окончания.
Мужчинам рекомендуется не мочиться в течение 3 часов (15–20 минут в случае обильных уретральных выделений) до взятия материала.
Информацию проверил
врач-эксперт

Информацию проверил врач-эксперт
Екатерина Демьяновская
Врач-невролог, кандидат медицинских наук
Оцените статью:
Полезная статья? Поделитесь в социальных сетях:
ВАЖНО
Информация из данного раздела не может служить достаточным основанием для постановки диагноза или назначения лечения. Решение об этом должен принимать врач на основании всех имеющихся у него данных.
Вам может быть интересно
Вам телеграм.
Telegram-канал,
которому, на наш взгляд,
можно доверять